.jpg)
Co to jest ReFacto AF?
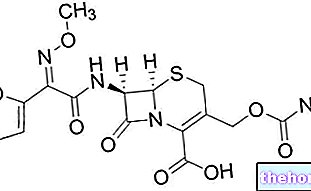
ReFacto AF składa się z proszku i rozpuszczalnika, które miesza się razem w celu uzyskania roztworu do wstrzykiwań. ReFacto AF zawiera substancję czynną moroktokog alfa.
Do czego służy ReFacto AF?
ReFacto AF stosuje się w leczeniu i zapobieganiu krwawieniom u pacjentów z hemofilią A (dziedziczne zaburzenie krwawienia). ReFacto AF można podawać pacjentom w każdym wieku, w tym noworodkom.
Lek jest dostępny wyłącznie na receptę.
Jak stosuje się ReFacto AF?
Terapię ReFacto AF powinien rozpoczynać lekarz doświadczony w leczeniu hemofilii A.
ReFacto AF podaje się we wstrzyknięciu dożylnym trwającym kilka minut. Dawka i częstość wstrzyknięć różni się w zależności od tego, czy ReFacto AF jest stosowany w leczeniu, zapobieganiu lub ograniczaniu krwawienia podczas zabiegu chirurgicznego. Dawkę należy dostosować w zależności od nasilenia i miejsce krwawienia lub rodzaj zabiegu. Wszystkie informacje na temat obliczania dawek znajdują się w ulotce dołączonej do opakowania.
Pacjenci lub opiekunowie mogą wykonywać zastrzyki ReFacto AF pod warunkiem, że otrzymali odpowiednie instrukcje.
Jak działa ReFacto AF?
Substancja czynna preparatu ReFacto AF, moroktokog alfa, to białkowy czynnik krzepnięcia krwi (substancja wspomagająca krzepnięcie krwi). Hemofilia A charakteryzuje się brakiem białka zwanego czynnikiem VIII, które bierze udział w krzepnięciu krwi.Niedobór czynnika VIII powoduje problemy z krzepnięciem krwi, takie jak krwawienie w stawach, mięśniach i narządach wewnętrznych.ReFacto AF, stosowany w celu uzupełnienia brakującego czynnika VIII umożliwia uzupełnienie niedoboru czynnika VIII i doraźną kontrolę zaburzeń krzepnięcia.
Moroktokog alfa nie jest ekstrahowany z ludzkiej krwi, ale jest wytwarzany metodą znaną jako „technologia rekombinacji DNA”: jest wytwarzany przez komórkę, która otrzymała gen (DNA), który umożliwia jej wytwarzanie ludzkiego czynnika VIII di krzepnięcia.
Jak badano ReFacto AF?
ReFacto AF został po raz pierwszy dopuszczony pod nazwą ReFacto w kwietniu 1999 r. do leczenia wcześniej leczonych i nieleczonych pacjentów z hemofilią A. Dopuszczenie to zostało oparte na wynikach trzech badań głównych.
W lutym 2009 roku wprowadzono kilka zmian w sposobie produkcji ReFacto, w tym wyeliminowano z procesu produkcyjnego stosowanie białka zwanego albuminą, które jest wytwarzane z ludzkiej krwi. Zmieniono również nazwę leku z ReFacto na ReFacto AF.
W następstwie tych zmian firma farmaceutyczna przeprowadziła badanie, aby wykazać, że organizm przyswaja ReFacto i ReFacto AF w ten sam sposób.Przeprowadziła również dwa główne badania dotyczące skuteczności preparatu ReFacto AF: pierwsze dotyczyło zapobiegania i leczenia epizodów krwawienia w 94 roku. wcześniej leczonych pacjentów i drugie leczenie krwawienia u 22 pacjentów poddawanych zabiegom chirurgicznym.
Jakie korzyści wykazał ReFacto AF podczas badań?
Badania wykazały, że ReFacto AF jest tak samo bezpieczny i skuteczny jak ReFacto w zapobieganiu i leczeniu epizodów krwawienia u pacjentów z hemofilią A.
Jakie jest ryzyko związane z ReFacto AF?
Pacjenci z hemofilią A mogą wytworzyć przeciwciała (inhibitory) przeciwko czynnikowi VIII. W takich przypadkach ReFacto AF nie jest skuteczny i tamowanie krwawienia może się nie powieść. Najczęstszym działaniem niepożądanym związanym ze stosowaniem leku ReFacto AF (obserwowanym u więcej niż 1 na 10 pacjentów) są wymioty.Pełny wykaz działań niepożądanych zgłaszanych podczas stosowania leku ReFacto AF znajduje się w ulotce dla pacjenta.
Leku ReFacto AF nie wolno podawać osobom, u których może występować nadwrażliwość (alergia) na ludzki czynnik krzepnięcia VIII, którąkolwiek inną substancję lub białka chomika.
Dlaczego ReFacto AF został zatwierdzony?
Komitet ds. Produktów Leczniczych Stosowanych u Ludzi (CHMP) zauważył, że ReFacto AF jest porównywalny z ReFacto, oryginalną postacią leku. CHMP uznał zatem, że korzyści płynące ze stosowania preparatu ReFacto AF przewyższają ryzyko w leczeniu i zapobieganiu krwawieniom u pacjentów z hemofilią A (wrodzonym niedoborem czynnika VIII). CHMP zalecił przyznanie pozwolenia na dopuszczenie do obrotu produktu ReFacto AF.
Jakie środki podejmuje się, aby zapewnić bezpieczne stosowanie ReFacto AF?
W związku ze stopniowym zastępowaniem ReFacto przez ReFacto AF na rynku producent leku dostarczy pakiety informacyjne pracownikom służby zdrowia, którzy będą przepisywać lub stosować ReFacto AF, wszystkim stowarzyszeniom pacjentów z hemofilią w Unii Europejskiej (UE) którzy przyjmują ReFacto AF oraz laboratoria, które będą monitorować pacjentów leczonych preparatem ReFacto AF Pakiety te będą zawierały informacje o różnicach między ReFacto i ReFacto AF, o bezpiecznym stosowaniu ReFacto AF, o sposobie zgłaszania działań niepożądanych, o podobnych lekach dostępnych poza UE i na koniec przypomina pacjentowi, aby na wypadek podróży miał ze sobą wystarczającą ilość leku ReFacto AF.
Inne informacje dotyczące ReFacto AF:
W dniu 13 kwietnia 1999 r. Komisja Europejska wydała „Pozwolenie na dopuszczenie do obrotu" dla ReFacto, ważne na terenie całej Unii Europejskiej. Posiadaczem „Pozwolenia na dopuszczenie do obrotu" dla produktu leczniczego jest Wyeth Europa Ltd. Dopuszczenie do obrotu przedłużono w dniu 13 kwietnia 2004 r. oraz 13 kwietnia 2009 r. W dniu 18 grudnia 2008 r. zmieniono nazwę leku na ReFacto AF.
Aby zobaczyć pełną wersję raportu ReFacto AF EPAR, kliknij tutaj.
Ostatnia aktualizacja niniejszego podsumowania: 04-2009.
Informacje o ReFacto AF - moroktokogu alfa opublikowane na tej stronie mogą być nieaktualne lub niekompletne. Aby prawidłowo wykorzystać te informacje, zobacz stronę Wyłączenie odpowiedzialności i przydatne informacje.