Składniki aktywne: Deferazyroks
EXJADE 125 mg tabletki do sporządzania zawiesiny doustnej
EXJADE 250 mg tabletki do sporządzania zawiesiny doustnej
EXJADE 500 mg tabletki do sporządzania zawiesiny doustnej
Ulotki informacyjne Exjade są dostępne dla wielkości opakowań: - EXJADE 125 mg tabletki do sporządzania zawiesiny doustnej, EXJADE 250 mg tabletki do sporządzania zawiesiny doustnej, EXJADE 500 mg tabletki do sporządzania zawiesiny doustnej
- EXJADE 90 mg tabletki powlekane, EXJADE 180 mg tabletki powlekane, EXJADE 360 mg tabletki powlekane
Dlaczego używa się Exjade? Po co to jest?
Co to jest EXJADE
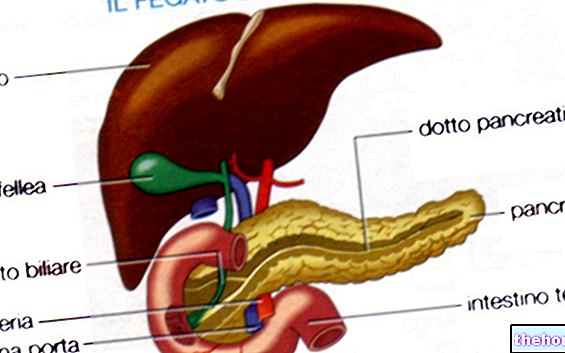
EXJADE zawiera substancję czynną o nazwie deferazyroks. Jest to chelator żelaza, czyli lek stosowany w celu usunięcia nadmiaru żelaza z organizmu (przeładowanie żelazem).Deferazyroks wiąże się z nadmiarem żelaza i usuwa go, eliminując go głównie z kałem.
Do czego służy EXJADE
U pacjentów z różnymi postaciami niedokrwistości (na przykład talasemia, anemia sierpowata lub zespoły mielodysplastyczne (MDS) mogą być wymagane wielokrotne transfuzje krwi. Mogą one jednak powodować nadmiar żelaza w organizmie. Dzieje się tak, ponieważ krew zawiera żelazo w organizmie nie ma naturalnego sposobu na usunięcie nadmiaru żelaza wprowadzonego przez transfuzje krwi U pacjentów z zespołami talasemii, którzy nie otrzymują transfuzji krwi, z czasem może się również rozwinąć przeładowanie żelazem, głównie z powodu podwyższonego ciśnienia krwi. zmniejszenie liczby krwinek. Z biegiem czasu nadmiar żelaza może uszkadzać ważne narządy, takie jak wątroba i serce. Leki zwane chelatorami żelaza służą do eliminacji żelaza.
EXJADE stosuje się w leczeniu obciążenia żelazem z powodu częstych transfuzji krwi u pacjentów z ciężką postacią talasemii beta w wieku 6 lat i starszych.
EXJADE stosuje się również w leczeniu przeładowania żelazem, gdy leczenie deferoksaminą jest przeciwwskazane lub niewystarczające u pacjentów z ciężką postacią talasemii beta z przeładowaniem żelazem spowodowanym rzadkimi transfuzjami krwi, u pacjentów z innymi postaciami niedokrwistości oraz u dzieci w wieku od 2 do 5 lat .
Preparat EXJADE stosuje się również, gdy terapia deferoksaminą jest przeciwwskazana lub niewystarczająca w leczeniu pacjentów w wieku 10 lat i starszych z obciążeniem żelazem związanym z zespołami talasemii, ale niewymagających transfuzji krwi.
Przeciwwskazania Kiedy nie należy stosować leku Exjade
Nie należy przyjmować leku EXJADE
- jeśli pacjent ma uczulenie na deferazyroks lub którykolwiek z pozostałych składników tego leku. Jeśli dotyczy to pacjenta, należy powiedzieć o tym lekarzowi przed przyjęciem leku EXJADE. Jeśli uważasz, że możesz być uczulony, poproś lekarza o poradę.
- jeśli u pacjenta występuje umiarkowana lub ciężka choroba nerek.
- jeśli pacjent obecnie przyjmuje jakiekolwiek inne leki chelatujące żelazo.
EXJADE nie jest zalecane
- jeśli pacjent znajduje się w zaawansowanym stadium zespołu mielodysplastycznego (MDS: zmniejszone wytwarzanie krwinek przez szpik kostny) lub ma zaawansowanego raka.
Środki ostrożności dotyczące stosowania Informacje ważne przed przyjęciem leku Exjade
Przed rozpoczęciem stosowania leku EXJADE należy omówić to z lekarzem lub farmaceutą:
- jeśli masz problemy z nerkami lub wątrobą.
- jeśli masz problemy z sercem z powodu przeładowania żelazem.
- jeśli pacjent zauważy znaczne zmniejszenie ilości wydalanego moczu (objaw problemów z nerkami).
- jeśli u pacjenta wystąpi ciężka wysypka skórna lub trudności w oddychaniu oraz zawroty głowy lub obrzęk, zwłaszcza twarzy i gardła (objawy ciężkiej reakcji alergicznej).
- jeśli u pacjenta występuje wysypka, zaczerwienienie skóry, pęcherze na wargach, oczach lub ustach, łuszczenie się skóry, gorączka (objawy ciężkiej reakcji skórnej)
- jeśli u pacjenta wystąpi jednocześnie senność, ból w prawym nadbrzuszu, zażółcenie lub nasilone zażółcenie skóry lub oczu oraz ciemne zabarwienie moczu (objawy problemów z wątrobą).
- jeśli wymiotujesz krwią i (lub) masz czarne stolce.
- jeśli u pacjenta występują częste bóle brzucha, szczególnie po jedzeniu lub zażyciu leku EXJADE.
- jeśli wystąpi częsta zgaga.
- jeśli w badaniach krwi występuje niski poziom płytek krwi lub białych krwinek.
- jeśli masz niewyraźne widzenie.
- jeśli masz biegunkę lub wymioty.
Jeśli którekolwiek z powyższych dotyczy Ciebie, natychmiast poinformuj o tym lekarza.
Monitorowanie leczenia za pomocą EXJADE
Podczas leczenia będziesz mieć regularne badania krwi i moczu. Sprawdzą ilość żelaza w organizmie (poziom ferrytyny we krwi), aby zobaczyć, jak działa lek EXJADE. Testy sprawdzą również czynność nerek (poziom kreatyniny we krwi, obecność białka w moczu) oraz czynność wątroby (poziom transaminaz we krwi). Lekarz może poprosić o biopsję nerki, jeśli podejrzewa znaczne uszkodzenie nerek. Wątrobę można również poddać testom rezonansu magnetycznego (MRI) w celu określenia ilości żelaza w wątrobie. Lekarz oceni te badania, aby zdecydować, która dawka leku EXJADE jest najbardziej odpowiednia dla pacjenta, a także użyje tych badań, aby zdecydować, kiedy przerwać stosowanie leku EXJADE.
W ramach środków ostrożności co roku podczas leczenia będą Państwo badani wzrok i słuch.
Interakcje Jakie leki lub pokarmy mogą zmienić działanie leku Exjade
Należy powiedzieć lekarzowi lub farmaceucie, jeśli pacjent przyjmuje, ostatnio przyjmował lub może przyjmować jakiekolwiek inne leki. Dotyczy to w szczególności:
- inne chelatory żelaza, których nie należy przyjmować z lekiem EXJADE,
- leki zobojętniające sok żołądkowy (leki stosowane w leczeniu zgagi) zawierające glin, których nie wolno przyjmować o tej samej porze dnia co lek EXJADE,
- cyklosporyna (stosowana w zapobieganiu odrzuceniu przeszczepionego narządu lub w innych stanach, takich jak reumatoidalne zapalenie stawów lub atopowe zapalenie skóry),
- symwastatyna (stosowana w celu obniżenia stężenia cholesterolu),
- niektóre leki przeciwbólowe lub przeciwzapalne (np. aspiryna, ibuprofen, kortykosteroidy),
- bisfosfoniany doustne (stosowane w leczeniu osteoporozy),
- leki przeciwzakrzepowe (stosowane w zapobieganiu lub leczeniu krzepnięcia krwi),
- hormonalne środki antykoncepcyjne (leki antykoncepcyjne),
- beprydyl, ergotamina (stosowane w chorobach serca i migrenach),
- repaglinid (stosowany w leczeniu cukrzycy),
- ryfampicyna (stosowana w leczeniu gruźlicy),
- fenytoina, fenobarbital, karbamazepina (stosowane w leczeniu padaczki),
- rytonawir (stosowany w leczeniu zakażenia wirusem HIV),
- paklitaksel (stosowany w leczeniu raka),
- teofilina (stosowana w leczeniu chorób układu oddechowego, takich jak astma),
- klozapina (stosowana w leczeniu zaburzeń psychicznych, takich jak schizofrenia)
- tyzanidyna (stosowana jako środek zwiotczający mięśnie),
- cholestyramina (stosowana w celu obniżenia poziomu cholesterolu we krwi).
Konieczne mogą być dodatkowe badania w celu monitorowania stężenia niektórych z tych leków we krwi.
Ostrzeżenia Ważne jest, aby wiedzieć, że:
Osoby w podeszłym wieku (65 lat lub starsze)
EXJADE może być stosowany przez osoby w wieku 65 lat i starsze w tej samej dawce, co u dorosłych. Pacjenci w podeszłym wieku mogą odczuwać więcej działań niepożądanych (zwłaszcza biegunki) niż pacjenci młodsi. Powinny być one ściśle monitorowane przez lekarza pod kątem działań niepożądanych, które mogą wymagać dostosowania dawki.
Dzieci i młodzież
EXJADE może być stosowany u dzieci i młodzieży w wieku 2 lat i starszych otrzymujących regularne transfuzje krwi oraz u dzieci i młodzieży w wieku 10 lat i starszych, które nie otrzymują regularnych transfuzji krwi. Lekarz dostosuje dawkę w zależności od wzrostu pacjenta
EXJADE nie jest zalecany dla dzieci poniżej 2 roku życia.
Ciąża i karmienie piersią
Jeśli pacjentka jest w ciąży lub karmi piersią, podejrzewa, że może być w ciąży lub planuje mieć dziecko, przed zastosowaniem tego leku należy zasięgnąć porady lekarza.
Lek EXJADE nie jest zalecany w okresie ciąży, chyba że jest to bezwzględnie konieczne.
Jeśli pacjentka obecnie stosuje doustny środek antykoncepcyjny lub plaster antykoncepcyjny w celu zapobiegania ciąży, musi stosować dodatkowy lub inny rodzaj antykoncepcji (np. prezerwatywy), ponieważ EXJADE może zmniejszać skuteczność doustnych i plastrów antykoncepcyjnych.
Nie zaleca się karmienia piersią podczas leczenia lekiem EXJADE.
Prowadzenie i używanie maszyn
W przypadku wystąpienia zawrotów głowy po przyjęciu leku EXJADE, nie należy prowadzić pojazdów ani obsługiwać żadnych narzędzi ani maszyn do czasu ustąpienia zawrotów głowy.
EXJADE zawiera laktozę
Jeśli pacjent został poinformowany przez lekarza, że nie toleruje niektórych cukrów, powinien skontaktować się z lekarzem przed przyjęciem tego produktu leczniczego.
Dawka, sposób i czas podawania Jak stosować Exjade: dawkowanie
Leczenie preparatem EXJADE będzie nadzorowane przez lekarza mającego doświadczenie w leczeniu przeładowania żelazem spowodowanego transfuzjami krwi.
Lek należy zawsze stosować zgodnie z zaleceniami lekarza. W razie wątpliwości skonsultuj się z lekarzem lub farmaceutą.
Ile EXJADE wziąć?
- U wszystkich pacjentów dawka produktu EXJADE jest uzależniona od masy ciała. Lekarz obliczy potrzebną dawkę i powie ile tabletek należy przyjmować każdego dnia.
- Zazwyczaj stosowana dawka dobowa leku EXJADE tabletki do sporządzania zawiesiny doustnej na początku leczenia u pacjentów otrzymujących regularne transfuzje krwi wynosi 20 mg na kilogram masy ciała Lekarz może zalecić większą lub mniejszą dawkę początkową w zależności od potrzeby indywidualnego leczenia.
- Zwykle dawka dobowa leku EXJADE tabletki do sporządzania zawiesiny doustnej na początku leczenia u pacjentów, którzy nie otrzymują regularnych transfuzji krwi, wynosi 10 mg na kilogram masy ciała.
- W zależności od odpowiedzi pacjenta na leczenie, lekarz może następnie zmodyfikować leczenie, zwiększając lub zmniejszając dawkę.
- Maksymalna zalecana dawka dobowa produktu leczniczego EXJADE tabletki do sporządzania zawiesiny doustnej wynosi 40 mg na kilogram masy ciała dla pacjentów otrzymujących regularne transfuzje krwi, 20 mg na kilogram masy ciała dla dorosłych pacjentów nieotrzymujących regularnych transfuzji krwi i 10 mg na kilogram masy ciała dla dzieci i młodzież, która nie otrzymuje regularnych transfuzji krwi.
Deferazyroks jest również dostępny w postaci „powlekanych” tabletek. W przypadku zmiany z tabletek powlekanych na te tabletki do sporządzania zawiesiny doustnej konieczne będzie dostosowanie dawki.
Kiedy stosować lek EXJADE?
- Lek EXJADE należy przyjmować raz na dobę, codziennie, mniej więcej o tej samej porze każdego dnia.
- Lek EXJADE tabletki do sporządzania zawiesiny doustnej należy przyjmować na pusty żołądek.
- Więc odczekaj co najmniej 30 minut przed zjedzeniem jakiegokolwiek jedzenia. Przyjmowanie leku EXJADE o tej samej porze każdego dnia pomoże również zapamiętać, kiedy należy przyjmować tabletki.
Jak przyjmować lek EXJADE?
- Wrzuć tabletkę (tabletki) do szklanki wody lub soku jabłkowego lub pomarańczowego (100-200 ml).
- Mieszaj, aż tabletka (tabletki) całkowicie się rozpuszczą. W szklance płyn będzie mętny.
- Wypij całą zawartość szklanki. Następnie dodaj trochę wody lub soku do pozostałości w szklance, wymieszaj i wypij ponownie.
Nie rozpuszczaj tabletek w napojach gazowanych lub mleku. Tabletek nie należy żuć, dzielić ani łamać. Nie należy połykać tabletek tak, jak są.
Jak długo należy przyjmować lek EXJADE?
Lek EXJADE należy przyjmować codziennie, tak długo, jak zaleci to lekarz. Jest to leczenie długoterminowe, które może trwać miesiące lub lata. Choroba będzie regularnie kontrolowana przez lekarza w celu sprawdzenia skuteczności leczenia (patrz także punkt 2: „Monitorowanie leczenia lekiem EXJADE”).
W przypadku jakichkolwiek pytań dotyczących czasu stosowania leku EXJADE, należy porozmawiać z lekarzem.
Pominięcie przyjęcia leku EXJADE
Jeśli zapomnisz przyjąć dawkę, weź ją tak szybko, jak sobie przypomnisz w tym dniu. Następną dawkę należy przyjąć zgodnie z planem. Nie należy przyjmować podwójnej dawki następnego dnia w celu uzupełnienia pominiętej tabletki (tabletek).
Przerwanie stosowania leku EXJADE
Nie należy przerywać stosowania leku EXJADE, chyba że tak zaleci lekarz. W przypadku przerwania jego stosowania nadmiar żelaza nie będzie już usuwany z organizmu (patrz również powyżej w punkcie „Jak długo stosować lek EXJADE”).
Przedawkowanie Co zrobić w przypadku przyjęcia zbyt dużej dawki leku Exjade
W przypadku zażycia zbyt dużej dawki leku EXJADE lub przypadkowego zażycia tabletek przez inną osobę, należy natychmiast skontaktować się z lekarzem lub szpitalem i pokazać im opakowanie tabletek, ponieważ może być konieczne leczenie.
Skutki uboczne Jakie są skutki uboczne leku Exjade
Jak każdy lek, lek ten może powodować działania niepożądane, chociaż nie u każdego one wystąpią. Większość działań niepożądanych ma nasilenie łagodne do umiarkowanego i zwykle ustępuje po okresie leczenia trwającym od kilku dni do kilku tygodni.
Niektóre działania niepożądane mogą być poważne i wymagać natychmiastowej pomocy lekarskiej.
Te działania niepożądane występują niezbyt często (mogą wystąpić u 1 na 100 osób) lub rzadko (mogą wystąpić u 1 na 1000 osób).
- Jeśli u pacjenta wystąpi ciężka wysypka skórna lub trudności w oddychaniu oraz zawroty głowy lub obrzęk, głównie twarzy i gardła (objawy ciężkiej reakcji alergicznej),
- jeśli u pacjenta występuje ciężka wysypka, zaczerwienienie skóry, pęcherze na wargach, oczach lub ustach, łuszczenie się skóry, gorączka (objawy ciężkiej reakcji skórnej),
- Jeśli zauważysz znaczne zmniejszenie ilości wydalanego moczu (objaw problemów z nerkami),
- jeśli u pacjenta wystąpi jednocześnie senność, ból w prawym nadbrzuszu, zażółcenie lub nasilone zażółcenie skóry lub oczu i ciemne zabarwienie moczu (objawy problemów z wątrobą),
- Jeśli wymiotujesz krwią i/lub masz czarne stolce,
- Jeśli u pacjenta często występują bóle brzucha, szczególnie po jedzeniu lub przyjmowaniu leku EXJADE,
- Jeśli doświadczasz częstej zgagi,
- Jeśli doświadczysz częściowej utraty wzroku,
- W przypadku wystąpienia silnego bólu w nadbrzuszu (zapalenie trzustki) należy przerwać stosowanie tego leku i natychmiast powiadomić lekarza.
Niektóre skutki uboczne mogą stać się poważne.
Te działania niepożądane są rzadkie.
- Jeśli masz niewyraźne lub niewyraźne widzenie,
- Jeśli masz ubytek słuchu,
jak najszybciej poinformować o tym lekarza.
Inne skutki uboczne
Bardzo często (może dotyczyć więcej niż 1 na 10 osób)
- Zmiany w testach czynności nerek.
Często (może dotyczyć do 1 na 10 osób)
- Zaburzenia żołądka i jelit, takie jak nudności, wymioty, biegunka, ból brzucha, wzdęcia, zaparcia, niestrawność
- Wysypka
- Bół głowy
- Nieprawidłowe testy czynności wątroby
- Swędzący
- Nieprawidłowy test moczu (białko w moczu)
Jeśli którykolwiek z tych objawów dotyczy cię poważnie, poinformuj o tym lekarza.
Niezbyt często (może dotyczyć do 1 na 100 osób)
- Zawroty głowy
- Gorączka
- Ból gardła
- Obrzęk rąk lub nóg
- przebarwienia skóry
- Lęk
- Zaburzenia snu
- Zmęczenie
Jeśli którykolwiek z tych objawów wystąpi poważnie, należy poinformować o tym lekarza.
Częstość nieznana (częstość nie może być określona na podstawie dostępnych danych).
- Zmniejszenie liczby komórek biorących udział w krzepnięciu krwi (małopłytkowość), liczby czerwonych krwinek (pogorszenie niedokrwistości), liczby białych krwinek (neutropenia) lub liczby wszystkich rodzajów krwinek ( pancytopenia)
- Wypadanie włosów
- Kamienie nerkowe
- Niski wydalanie moczu
- Rozdarcie w ścianie żołądka lub jelit, które może być bolesne i powodować nudności
- Silny ból w górnej części brzucha (zapalenie trzustki)
- Zwiększona kwasowość krwi (kwasica metaboliczna).
Zgłaszanie skutków ubocznych
Jeśli wystąpią jakiekolwiek objawy niepożądane, w tym wszelkie możliwe działania niepożądane niewymienione w tej ulotce, należy porozmawiać z lekarzem lub farmaceutą. Możesz również zgłaszać skutki uboczne bezpośrednio za pośrednictwem krajowego systemu raportowania. Zgłaszając działania niepożądane, możesz dostarczyć więcej informacji na temat bezpieczeństwa tego leku.
Wygaśnięcie i przechowywanie
- Lek należy przechowywać w miejscu niewidocznym i niedostępnym dla dzieci.
- Nie stosować tego leku po upływie terminu ważności zamieszczonego na blistrze po EXP i kartoniku po EXP.Termin ważności oznacza ostatni dzień podanego miesiąca.
- Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.
- Nie używać opakowań uszkodzonych lub noszących ślady ingerencji.
- Nie należy wyrzucać żadnych leków do kanalizacji ani domowych pojemników na odpadki. Należy zapytać farmaceutę, jak usunąć leki, których się już nie używa.Pomoże to chronić środowisko.
Inne informacje
Co zawiera lek EXJADE
Substancją czynną jest deferazyroks.
Każda tabletka do sporządzania zawiesiny doustnej EXJADE 125 mg zawiera 125 mg deferazyroksu.
Każda tabletka do sporządzania zawiesiny doustnej EXJADE 250 mg zawiera 250 mg deferazyroksu.
Każda tabletka do sporządzania zawiesiny doustnej EXJADE 500 mg zawiera 500 mg deferazyroksu.
Pozostałe składniki to: laktoza jednowodna, krospowidon typu A, powidon, laurylosiarczan sodu, celuloza mikrokrystaliczna, krzemionka koloidalna bezwodna i magnezu stearynian.
Opis jak wygląda EXJADE i zawartość opakowania
EXJADE jest dostarczany w postaci tabletek do sporządzania zawiesiny. Tabletki są białawe, okrągłe i płaskie.
- Tabletki EXJADE 125 mg są oznaczone „J 125” po jednej stronie i „NVR” po drugiej.
- Tabletki EXJADE 250 mg są oznaczone „J 250” po jednej stronie i „NVR” po drugiej.
- Tabletki EXJADE 500 mg są oznaczone „J 500” po jednej stronie i „NVR” po drugiej.
EXJADE 125 mg, 250 mg i 500 mg tabletki do sporządzania zawiesiny doustnej są dostępne w opakowaniach jednostkowych zawierających 28, 84 lub 252 tabletki do sporządzania zawiesiny doustnej.
EXJADE 500 mg tabletki do sporządzania zawiesiny doustnej są również dostępne w opakowaniach zbiorczych zawierających 294 (3 opakowania po 98) tabletek do sporządzania zawiesiny.
Nie wszystkie wielkości lub moce opakowań mogą być sprzedawane w Twoim kraju.
Ulotka pakietu źródłowego: AIFA (Włoska Agencja Leków). Treść opublikowana w styczniu 2016 r. Przedstawione informacje mogą być nieaktualne.
Aby mieć dostęp do najbardziej aktualnej wersji, warto wejść na stronę AIFA (Włoskiej Agencji Leków). Zastrzeżenie i przydatne informacje.
01.0 NAZWA PRODUKTU LECZNICZEGO
EXJADE 125 MG TABLETKI DO DYSPRESJI
▼ Produkt leczniczy podlegający dodatkowemu monitorowaniu. Umożliwi to szybką identyfikację nowych informacji dotyczących bezpieczeństwa. Osoby należące do fachowego personelu medycznego proszone są o zgłaszanie wszelkich podejrzewanych działań niepożądanych. Patrz punkt 4.8, aby uzyskać informacje na temat zgłaszania działań niepożądanych.
02.0 SKŁAD JAKOŚCIOWY I ILOŚCIOWY
Każda tabletka do sporządzania zawiesiny zawiera 125 mg deferazyroksu.
Zaróbka:
Każda tabletka do sporządzania zawiesiny zawiera 136 mg laktozy.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
03.0 POSTAĆ FARMACEUTYCZNA
Tabletka dyspergowalna
Białawe, okrągłe, płaskie tabletki o ściętych krawędziach, z wytłoczonym oznaczeniem NVR po jednej stronie i J 125 po drugiej.
04.0 INFORMACJE KLINICZNE
04.1 Wskazania terapeutyczne
EXJADE jest wskazany w leczeniu przewlekłego obciążenia żelazem w wyniku częstych transfuzji krwi (≥7 ml/kg mc./miesiąc stężonych krwinek czerwonych) u pacjentów z ciężką postacią talasemii beta w wieku 6 lat i starszych.
EXJADE jest również wskazany w leczeniu przewlekłego obciążenia żelazem w wyniku przetoczeń krwi, gdy leczenie deferoksaminą jest przeciwwskazane lub niewystarczające w następujących grupach pacjentów:
- u pacjentów z ciężką postacią talasemii beta z obciążeniem żelazem z powodu częstych transfuzji krwi (≥7 ml/kg/miesiąc stężonych krwinek czerwonych) w wieku od 2 do 5 lat,
- u pacjentów z ciężką postacią talasemii beta z obciążeniem żelazem z powodu rzadkich transfuzji krwi (
- u pacjentów z inną niedokrwistością w wieku 2 lat i starszych.
EXJADE jest również wskazany w leczeniu przewlekłego obciążenia żelazem, wymagającego leczenia chelatującego, gdy terapia deferoksaminą jest przeciwwskazana lub nieodpowiednia u pacjentów z zespołami talasemii niezależnymi od transfuzji krwi w wieku 10 lat i starszymi.
04.2 Dawkowanie i sposób podawania
Leczenie preparatem EXJADE powinni rozpoczynać i prowadzić lekarze doświadczeni w leczeniu przewlekłego obciążenia żelazem.
Dawkowanie – przeładowanie żelazem z powodu transfuzji krwi
Zaleca się rozpoczęcie leczenia po przetoczeniu około 20 jednostek (około 100 ml/kg) stężonych krwinek czerwonych lub gdy monitorowanie kliniczne wykazuje obecność przewlekłego obciążenia żelazem (np. stężenie ferrytyny w surowicy > 1000 mcg/l). Dawki (w mg/kg) należy obliczyć i zaokrąglić do najbliższej pełnej tabletki.
Celem terapii chelatacją żelaza jest wyeliminowanie ilości żelaza podawanego podczas transfuzji oraz, w razie potrzeby, zmniejszenie istniejącego ładunku żelaza.
Dawka początkowa
Zalecana początkowa dawka dobowa leku EXJADE wynosi 20 mg/kg masy ciała.
Można rozważyć początkową dawkę dobową 30 mg/kg u pacjentów, którzy muszą zmniejszyć wysoki poziom żelaza w organizmie i którzy otrzymują również ponad 14 ml/kg/miesiąc skoncentrowanych krwinek czerwonych (około > 4 jednostki/miesiąc w przypadku osoby dorosłej). ).
Można rozważyć początkową dawkę dobową 10 mg/kg mc. u pacjentów, którzy nie muszą zmniejszać poziomu żelaza w organizmie i którzy otrzymują również mniej niż 7 ml/kg/miesiąc skoncentrowanych krwinek czerwonych (w przybliżeniu
W przypadku pacjentów już odpowiednio leczonych deferoksaminą można rozważyć początkową dawkę leku EXJADE, która jest liczbowo równa połowie dawki deferoksaminy (np. pacjent otrzymujący 40 mg/kg mc./dobę deferoksaminy przez 5 dni w tygodniu (lub odpowiednik) może przejść na początkowa dawka dobowa 20 mg/kg/dobę preparatu EXJADE). Gdy skutkuje to dobową dawką mniejszą niż 20 mg/kg masy ciała, odpowiedź pacjenta należy monitorować, a jeśli nie zostanie osiągnięta wystarczająca skuteczność, należy rozważyć zwiększenie dawki (patrz punkt 5.1).
Dostosowanie dawki
Zaleca się comiesięczne monitorowanie stężenia ferrytyny w surowicy i, w razie potrzeby, dostosowanie dawki preparatu EXJADE co 3-6 miesięcy, na podstawie trendu stężenia ferrytyny w surowicy.Dostosowanie dawki można dokonywać w odstępach od 5 do 10 mg/kg powinien być dostosowany do indywidualnej odpowiedzi pacjenta i celów terapeutycznych (utrzymanie lub zmniejszenie obciążenia żelazem). można rozważyć dawki do 40 mg/kg.Dostępność danych dotyczących długoterminowej skuteczności i bezpieczeństwa stosowania preparatu EXJADE w dawkach powyżej 30 mg/kg jest obecnie ograniczona (264 pacjentów obserwowanych średnio przez 1 rok po zwiększeniu dawki). bardzo słabą kontrolę hemosyderozy osiąga się przy dawkach do 30 mg/kg, dalszy wzrost (maksymalnie i 40 mg / kg) może nie zapewnić zadowalającej kontroli i można rozważyć alternatywne opcje leczenia. Jeżeli przy dawkach powyżej 30 mg/kg nie zostanie osiągnięta zadowalająca kontrola, leczenie tymi dawkami nie powinno być kontynuowane i w miarę możliwości należy rozważyć alternatywne opcje leczenia. Dawki powyżej 40 mg/kg nie są zalecane, ponieważ doświadczenie z dawkami powyżej tego poziomu jest ograniczone.
U pacjentów leczonych dawkami powyżej 30 mg/kg należy rozważyć zmniejszenie dawki w zakresie od 5 do 10 mg/kg po osiągnięciu kontroli (np. stężenie ferrytyny w surowicy utrzymujące się poniżej 2500 mcg/l, które wykazuje tendencję spadkową w czasie). . U pacjentów, u których stężenie ferrytyny w surowicy osiągnęło wartość referencyjną (zwykle od 500 do 1000 mcg/l), należy rozważyć zmniejszenie dawki w zakresie od 5 do 10 mg/kg w celu utrzymania stężenia ferrytyny w surowicy w zakresie wartości referencyjnych. stale spada poniżej 500 mcg/l, należy rozważyć przerwanie leczenia (patrz punkt 4.4).
Dawkowanie - zespoły talasemii niezależne od transfuzji krwi
Terapię chelatującą należy rozpocząć tylko wtedy, gdy istnieją dowody na przeładowanie żelazem (stężenie żelaza w wątrobie (LIC) ≥5 mg Fe/g/dw lub stale stężenie ferrytyny w surowicy > 800 mcg/l). LIC jest najlepszą metodą określania przeładowania żelazem i powinna być stosowana wszędzie tam, gdzie jest dostępna. Należy zachować ostrożność u wszystkich pacjentów podczas terapii chelatacyjnej, aby zminimalizować ryzyko nadmiernej chelatacji.
Dawka początkowa
U pacjentów z zespołami talasemii niezależnymi od transfuzji krwi zalecana początkowa dawka dobowa produktu EXJADE wynosi 10 mg/kg masy ciała.
Dostosowanie dawki
Zaleca się comiesięczne monitorowanie stężenia ferrytyny w surowicy. Po każdych 3-6 miesiącach leczenia należy rozważyć zwiększanie dawki o 5 do 10 mg/kg, jeśli LIC pacjenta wynosi ≥7 mg Fe/g dw lub jeśli stężenie ferrytyny w surowicy stale wynosi > 2000 mcg/l i nie wykazuje tendencja spadkowa i jeśli pacjent dobrze toleruje lek. Dawki powyżej 20 mg/kg nie są zalecane, ponieważ nie ma doświadczenia z dawkami powyżej tego poziomu u pacjentów z zespołami talasemii niezależnymi od transfuzji.
U pacjentów, u których nie przeprowadzono oceny LIC, a stężenie ferrytyny w surowicy wynosi ≤2000 mcg/l, dawka nie powinna przekraczać 10 mg/kg.
U pacjentów, których dawka została zwiększona powyżej 10 mg/kg, zaleca się zmniejszenie dawki do 10 mg/kg lub mniej, gdy LIC jest
Przerwanie leczenia
Leczenie należy przerwać po osiągnięciu zadowalającego poziomu żelaza w organizmie (LIC
Specjalne kategorie pacjentów
Pacjenci w podeszłym wieku (≥65 lat)
Zalecenia dotyczące dawkowania dla pacjentów w podeszłym wieku są takie same jak opisane powyżej. W badaniach klinicznych u pacjentów w podeszłym wieku działania niepożądane występowały częściej niż u młodszych pacjentów (w szczególności biegunka) i należy ich ściśle monitorować pod kątem działań niepożądanych, które mogą wymagać dostosowania dawki.
Populacja pediatryczna
Zalecenia dotyczące dawkowania u dzieci w wieku 2-17 lat z obciążeniem żelazem w wyniku transfuzji krwi są takie same jak u pacjentów dorosłych. Obliczenie dawki powinno uwzględniać zmiany masy ciała dzieci i młodzieży w czasie.
U dzieci z obciążeniem żelazem w wyniku transfuzji krwi w wieku od 2 do 5 lat ekspozycja jest mniejsza niż u dorosłych (patrz punkt 5.2).W rezultacie pacjenci w tej grupie wiekowej mogą wymagać większych dawek niż jest to wymagane. u dorosłych, jednak dawka początkowa powinno być takie samo jak dla dorosłych, a następnie należy dostosować indywidualne.
U pacjentów pediatrycznych z zespołami talasemii niezależnymi od transfuzji krwi dawka nie powinna przekraczać 10 mg/kg. U tych pacjentów ściślejsza kontrola LIC i ferrytyny w surowicy jest niezbędna, aby uniknąć nadmiernej chelatacji: oprócz comiesięcznej oceny ferrytyny w surowicy, LIC należy sprawdzać co trzy miesiące, gdy ferrytyna w surowicy wynosi ≤800 mcg / l.
Nie ustalono bezpieczeństwa i skuteczności produktu EXJADE u niemowląt od urodzenia do 23 miesiąca życia.Brak dostępnych danych.
Pacjenci z zaburzeniami czynności nerek
Preparat EXJADE nie był badany u pacjentów z zaburzeniami czynności nerek i jest przeciwwskazany u pacjentów z oszacowanym klirensem kreatyniny.
Pacjenci z zaburzeniami czynności wątroby
Produkt EXJADE nie jest zalecany u pacjentów z ciężkimi zaburzeniami czynności wątroby (klasa C w skali Child-Pugh). U pacjentów z umiarkowanymi zaburzeniami czynności wątroby (klasa B wg Childa-Pugha) dawkę należy znacznie zmniejszyć, a następnie stopniowo zwiększać do granicy 50% (patrz punkty 4.4 i 5.2), a produkt leczniczy EXJADE należy stosować z ostrożnością. takich pacjentów. Czynność wątroby należy sprawdzać u wszystkich pacjentów przed leczeniem, co 2 tygodnie w pierwszym miesiącu, a następnie co miesiąc (patrz punkt 4.4).
Sposób podawania
Do stosowania doustnego.
Lek EXJADE należy przyjmować raz na dobę na pusty żołądek, co najmniej 30 minut przed posiłkiem, najlepiej codziennie o tej samej porze (patrz punkty 4.5 i 5.2).
Tabletki rozpuszcza się mieszając je w szklance wody lub soku pomarańczowego lub jabłkowego (100-200 ml), aż do uzyskania drobnej zawiesiny. Po spożyciu zawiesiny wszelkie pozostałości należy ponownie zawiesić w niewielkiej ilości wody lub soku i połknąć. Tabletek nie wolno żuć ani połykać w całości (patrz również punkt 6.2).
04.3 Przeciwwskazania
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Połączenie z innymi terapiami chelatującymi żelazo, ponieważ bezpieczeństwo tych połączeń nie zostało ustalone (patrz punkt 4.5).
Pacjenci z szacowanym klirensem kreatyniny
04.4 Specjalne ostrzeżenia i odpowiednie środki ostrożności dotyczące stosowania
Funkcja nerki:
Preparat EXJADE badano wyłącznie u pacjentów z wyjściowym stężeniem kreatyniny w surowicy w prawidłowym zakresie odpowiednim dla wieku.
Podczas badań klinicznych u około 36% pacjentów wystąpiło >33% zwiększenie stężenia kreatyniny w surowicy w ≥2 kolejnych przypadkach, czasami powyżej górnej granicy normy. Wzrost ten był zależny od dawki.U około dwóch trzecich pacjentów u pozostałych pacjentów wzrost stężenia kreatyniny w surowicy nie zawsze reagował na zmniejszenie dawki lub przerwanie leczenia Przypadki ostrej niewydolności nerek po wprowadzeniu produktu leczniczego EXJADE do obrotu (patrz punkt 4.8). W niektórych przypadkach po wprowadzeniu leku do obrotu pogorszenie czynności nerek prowadziło do niewydolności nerek wymagającej czasowej lub stałej dializy.
Przyczyny zwiększenia stężenia kreatyniny w surowicy nie zostały wyjaśnione, dlatego należy zwrócić szczególną uwagę na monitorowanie stężenia kreatyniny u pacjentów przyjmujących jednocześnie leki obniżające czynność nerek oraz u pacjentów otrzymujących duże dawki produktu leczniczego EXJADE i (lub) z małą częstością przetoczeń krwi. (
Zaleca się dwukrotną ocenę kreatyniny w surowicy przed rozpoczęciem leczenia. Poziom kreatyniny w surowicy, klirens kreatyniny (oszacowany za pomocą wzoru Cockcrofta-Gaulta lub MDRD u dorosłych i wzoru Schwartza u dzieci) i (lub) stężenie cystatyny C w osoczu należy monitorować co tydzień przez pierwszy miesiąc po rozpoczęciu lub modyfikacji leczenia produktem EXJADE i raz w miesiącu Należy zachować ostrożność w celu utrzymania odpowiedniego nawodnienia u pacjentów z biegunką lub wymiotami.
Po wprowadzeniu produktu leczniczego EXJADE do obrotu zgłaszano przypadki kwasicy metabolicznej. Większość z tych pacjentów miała zaburzenia czynności nerek, zaburzenia czynności kanalika nerkowego (zespół Fanconiego) lub biegunkę lub stany, w których zaburzenia równowagi kwasowo-zasadowej są znanym powikłaniem. W tych populacjach należy monitorować równowagę kwasowo-zasadową zgodnie ze wskazaniami klinicznymi. Należy rozważyć przerwanie przeszczepu produktu EXJADE u pacjentów, u których wystąpiła kwasica metaboliczna.
W przypadku pacjentów dorosłych dawkę dobową można zmniejszyć o 10 mg/kg, jeśli podczas dwóch kolejnych wizyt obserwuje się wzrost stężenia kreatyniny w surowicy o >33% powyżej średniej wartości sprzed leczenia i zmniejszenie klirensu. dolna granica normalnego zakresu (
Jeżeli po zmniejszeniu dawki u pacjentów dorosłych i dzieci obserwuje się wzrost stężenia kreatyniny w surowicy > 33% powyżej średniej wartości sprzed leczenia i (lub) obliczony klirens kreatyniny spada poniżej dolnej granicy „zakresu normalnego”, należy zastosować leczenie Leczenie można wznowić w zależności od indywidualnych okoliczności klinicznych.
Testy na białkomocz należy wykonywać co miesiąc. W razie potrzeby można również monitorować inne markery czynności kanalików nerkowych (np. cukromocz u pacjentów bez cukrzycy i niskie stężenie potasu, fosforanów, magnezu lub moczanu w surowicy, fosfaturia, aminoacyduria). Można rozważyć zmniejszenie dawki lub przerwanie leczenia w przypadku nieprawidłowości w poziomach markerów czynności kanalików i (lub) wskazań klinicznych. Tubulopatia nerkowa była zgłaszana głównie u dzieci i młodzieży z talasemią beta leczonych produktem EXJADE.
Jeżeli pomimo zmniejszenia dawki lub przerwania leczenia stężenie kreatyniny w surowicy pozostaje znacząco podwyższone i jeśli również występuje utrzymująca się nieprawidłowość w innym markerze czynności nerek (np. białkomocz, zespół Fanconiego), należy skierować pacjenta do nefrologa i wykonać dodatkowe badania specjalistyczne (takich jak biopsja nerki).
Funkcja wątroby:
U pacjentów leczonych produktem leczniczym EXJADE obserwowano podwyższenie wyników testów czynności wątroby. Po wprowadzeniu produktu do obrotu zgłaszano przypadki niewydolności wątroby, niektóre ze skutkiem śmiertelnym, u pacjentów leczonych produktem leczniczym EXJADE. Większość przypadków niewydolności wątroby dotyczyła pacjentów ze znaczną chorobowością, w tym z istniejącą wcześniej marskością wątroby. Nie można jednak wykluczyć roli produktu EXJADE jako czynnika przyczyniającego się lub nasilającego (patrz punkt 4.8).
Zaleca się sprawdzanie aktywności aminotransferaz w surowicy, stężenia bilirubiny i fosfatazy alkalicznej przed rozpoczęciem leczenia, co 2 tygodnie przez pierwszy miesiąc, a następnie co miesiąc. Po wyjaśnieniu przyczyny nieprawidłowości w testach czynności wątroby lub po powrocie do normalnych poziomów można rozważyć ostrożne wznowienie leczenia niższą dawką, a następnie stopniowe zwiększanie dawki.
Produkt leczniczy EXJADE nie jest zalecany u pacjentów z ciężkimi zaburzeniami czynności wątroby (klasa C w skali Child-Pugh) (patrz punkt 5.2).
Podsumowanie zaleceń dotyczących monitorowania bezpieczeństwa:
U pacjentów z krótką przewidywaną długością życia (np. z zespołami miolodysplastycznymi wysokiego ryzyka), szczególnie gdy współistniejąca zachorowalność może zwiększać ryzyko działań niepożądanych, korzyści ze stosowania preparatu EXJADE mogą być ograniczone i mniejsze niż ryzyko. W związku z tym leczenie produktem EXJADE nie jest zalecane u tych pacjentów.
Należy zachować ostrożność u pacjentów w podeszłym wieku ze względu na większą częstość występowania działań niepożądanych (w szczególności biegunki).
Dane dotyczące dzieci z talasemią niezależną od transfuzji krwi są bardzo ograniczone (patrz punkt 5.1). W związku z tym w populacji pediatrycznej leczenie produktem EXJADE należy bardzo dokładnie monitorować pod kątem działań niepożądanych i monitorować obciążenie żelazem. Ponadto przed rozpoczęciem leczenia dzieci z talasemią niezależną od transfuzji krwi i nadmiernym obciążeniem żelazem preparatem EXJADE lekarze powinni mieć świadomość, że konsekwencje długotrwałej ekspozycji u tych pacjentów nie są obecnie znane.
Zaburzenia żołądkowo-jelitowe
U pacjentów otrzymujących lek EXJADE, w tym u dzieci i młodzieży, zgłaszano owrzodzenie górnego odcinka przewodu pokarmowego i krwotoki. U niektórych pacjentów obserwowano mnogie owrzodzenia (patrz punkt 4.8). Zgłaszano przypadki powikłanych owrzodzeń z perforacją przewodu pokarmowego Zgłaszano również przypadki krwawień z przewodu pokarmowego prowadzące do zgonu, zwłaszcza u pacjentów w podeszłym wieku z nowotworami złośliwymi układu krwiotwórczego i (lub) małą liczbą płytek krwi.W trakcie leczenia produktem leczniczym EXJADE należy o tym poinformować lekarzy i pacjentów. zwracać uwagę na oznaki i objawy owrzodzenia i krwawienia z przewodu pokarmowego oraz niezwłocznie rozpocząć ocenę i leczenie skojarzone w przypadku podejrzenia wystąpienia poważnych działań niepożądanych ze strony przewodu pokarmowego. Należy zachować ostrożność u pacjentów przyjmujących lek EXJADE w skojarzeniu z substancjami o rozpoznanym potencjale wrzodowym, takimi jak niesteroidowe leki przeciwzapalne, kortykosteroidy lub doustne bisfosfoniany, u pacjentów leczonych lekami przeciwzakrzepowymi oraz u pacjentów z liczbą płytek krwi mniejszą niż 50 000 / mm3 (50 x 109 / l) (patrz rozdział 4.5).
Schorzenia skóry
Podczas leczenia lekiem EXJADE mogą pojawić się wysypki skórne. W większości przypadków wysypki ustępują samoistnie. Jeśli konieczne jest przerwanie leczenia, leczenie można wznowić po ustąpieniu wyprysku, stosując mniejszą dawkę, którą następnie można stopniowo zwiększać. W ciężkich przypadkach można wznowić leczenie w połączeniu z doustnym podaniem sterydów na krótki czas. Po wprowadzeniu produktu do obrotu zgłaszano przypadki zespołu Stevensa-Johnsona (SJS). Nie można wykluczyć ryzyka innych, poważniejszych reakcji skórnych [toksyczna nekroliza naskórka (TEN), reakcja na lek z eozynofilią i objawami ogólnoustrojowymi (DRESS)]. W przypadku podejrzenia SJS lub jakiejkolwiek innej poważnej reakcji skórnej należy natychmiast przerwać stosowanie preparatu EXJADE i nie wolno go ponownie wprowadzać.
Reakcje nadwrażliwości
U pacjentów otrzymujących preparat EXJADE zgłaszano przypadki ciężkich reakcji nadwrażliwości (takich jak anafilaksja i obrzęk naczynioruchowy), które w większości przypadków wystąpiły w ciągu pierwszego miesiąca leczenia (patrz punkt 4.8). W przypadku wystąpienia takich reakcji należy przerwać stosowanie produktu EXJADE i wdrożyć odpowiednią interwencję medyczną. Ze względu na ryzyko wstrząsu anafilaktycznego, deferazyroksu nie należy ponownie stosować u pacjentów, u których wystąpiła reakcja nadwrażliwości (patrz punkt 4.3).
Wzrok i słuch
Zgłaszano zaburzenia słuchu (utrata słuchu) i oczu (zmętnienie soczewki) (patrz punkt 4.8).Zaleca się wykonanie badań słuchu i okulisty (w tym badania dna oka) przed rozpoczęciem leczenia, a następnie w regularnych odstępach czasu (co 12 miesięcy). . Jeśli podczas leczenia wystąpią zaburzenia, można rozważyć zmniejszenie dawki lub przerwanie leczenia.
Choroby krwi
U pacjentów leczonych produktem leczniczym EXJADE zgłaszano przypadki leukopenii, trombocytopenii lub pancytopenii (lub nasilenia tych cytopenii) oraz nasilenia niedokrwistości. nie można wykluczyć przyczynowej lub nasilającej roli leczenia. Należy rozważyć przerwanie leczenia u pacjentów, u których rozwinie się cytopenia, której nie można przypisać żadnej przyczynie.
Inne względy
Zaleca się comiesięczne monitorowanie stężenia ferrytyny w surowicy w celu oceny odpowiedzi pacjenta na leczenie (patrz punkt 4.2). Jeśli poziom ferrytyny w surowicy stale spada poniżej 500 mcg/l (przy obciążeniu żelazem z powodu przetoczeń krwi) lub poniżej 300 mcg/l (w zespołach talasemii niezależnych od transfuzji), należy rozważyć możliwość „przerwania leczenia”.
Wyniki badań stężenia kreatyniny, ferrytyny w surowicy i transaminaz w surowicy powinny być regularnie rejestrowane i oceniane w celu monitorowania ich postępów, a także należy je zapisywać w zeszycie dostarczonym pacjentowi.
W badaniu klinicznym leczenie produktem EXJADE przez okres do 5 lat nie wpływało na wzrost i rozwój płciowy leczonych pacjentów pediatrycznych. Jednakże, jako ogólny środek ostrożności w postępowaniu z pacjentami pediatrycznymi z obciążeniem żelazem spowodowanym transfuzjami krwi, należy w regularnych odstępach czasu (co 12 miesięcy) monitorować masę ciała, wzrost i rozwój płciowy.
Dysfunkcja serca jest znanym powikłaniem ciężkiego przeładowania żelazem. U pacjentów z ciężkim obciążeniem żelazem podczas długotrwałego leczenia produktem EXJADE należy monitorować czynność serca.
Każda tabletka zawiera 136 mg laktozy. Pacjenci z rzadką dziedziczną nietolerancją galaktozy, niedoborem laktazy typu Lapp, zespołem złego wchłaniania glukozy-galaktozy lub ciężkim niedoborem laktazy nie powinni przyjmować tego leku.
04.5 Interakcje z innymi produktami leczniczymi i inne formy interakcji
Bezpieczeństwo preparatu EXJADE w połączeniu z innymi chelatorami żelaza nie zostało ustalone. Dlatego nie należy go łączyć z innymi terapiami chelatującymi żelazo (patrz punkt 4.3).
Jednoczesne podawanie produktu EXJADE z substancjami o uznanym działaniu wrzodowym, takimi jak niesteroidowe leki przeciwzapalne (w tym kwas acetylosalicylowy w dużych dawkach), doustne kortykosteroidy lub bisfosfoniany może zwiększać ryzyko toksycznego działania na przewód pokarmowy (patrz punkt 4.4). EXJADE z lekami przeciwzakrzepowymi może również zwiększać ryzyko krwotoku z przewodu pokarmowego W przypadku skojarzenia deferazyroksu z tymi substancjami wymagana jest ścisła obserwacja kliniczna.
Biodostępność deferazyroksu była zmiennie zwiększana podczas jednoczesnego przyjmowania pokarmu, dlatego produkt EXJADE należy przyjmować na pusty żołądek, co najmniej 30 minut przed posiłkiem, najlepiej o tej samej porze każdego dnia (patrz punkty 4.2 i 5.2).
Deferazyroks jest metabolizowany przez enzymy UGT. W badaniu z udziałem zdrowych ochotników jednoczesne podawanie produktu EXJADE (pojedyncza dawka 30 mg/kg) i silnego induktora enzymu UGT ryfampicyny (powtarzana dawka 600 mg/dobę) spowodowało zmniejszenie ekspozycji na deferazyroks o 44% (90 % CI: 37% - 51%). Dlatego jednoczesne stosowanie produktu EXJADE z silnymi induktorami enzymów UGT (np. ryfampicyną, karbamazepiną, fenytoiną, fenobarbitalem, rytonawirem) może prowadzić do zmniejszenia skuteczności produktu EXJADE.W trakcie i po jednoczesnym leczeniu należy monitorować stężenie ferrytyny w surowicy pacjenta i, jeśli to konieczne, dostosować dawkę leku EXJADE.
W badaniu mechanistycznym mającym na celu określenie stopnia recyrkulacji jelitowo-wątrobowej cholestyramina znacząco zmniejszała ekspozycję na deferazyroks (patrz punkt 5.2).
W badaniu z udziałem zdrowych ochotników jednoczesne podawanie produktu EXJADE i midazolamu (substratu cytochromu CYP3A4) powodowało zmniejszenie ekspozycji na midazolam o 17% (90% CI: 8% - 26%). należy stosować, gdy deferazyroks jest łączony z lekami metabolizowanymi przez CYP3A4 (np. cyklosporyną, simwastatyną, hormonalnymi środkami antykoncepcyjnymi, beprydylem, ergotaminą) ze względu na możliwe zmniejszenie ich skuteczności.
W badaniu z udziałem zdrowych ochotników jednoczesne podawanie deferazyroksu jako umiarkowanego inhibitora CYP2C8 (30 mg/kg mc./dobę) z repaglinidem będącym substratem CYP2C8, podawanym w pojedynczej dawce 0,5 mg, zwiększyło AUC i Cmax repaglinidu około 2,3 razy ( 90% CI [2,03–2,63]) i 1,6 razy (90% CI [1,42–1,84]) dla repaglinidu większego niż 0,5 mg nie zostało określone, należy unikać jednoczesnego stosowania deferazyroksu z repaglinidem. należy monitorować stężenie glukozy, jeśli takie połączenie wydaje się konieczne (patrz punkt 4.4). Nie można wykluczyć interakcji między deferazyroksem a innymi substratami CYP2C8, takimi jak paklitaksel.
W badaniu z udziałem zdrowych ochotników jednoczesne podawanie produktu EXJADE jako inhibitora CYP1A2 (powtarzana dawka 30 mg/kg/dobę) i teofiliny będącej substratem CYP1A2 (pojedyncza dawka 120 mg) powodowało zwiększenie AUC 84% teofiliny (90 % CI: 73% -95%). Cmax pojedynczej dawki nie uległo zmianie, ale przy długotrwałym podawaniu oczekuje się wzrostu Cmax teofiliny. Dlatego nie zaleca się jednoczesnego stosowania produktu EXJADE i teofiliny.W przypadku jednoczesnego stosowania produktu EXJADE i teofiliny należy rozważyć monitorowanie stężenia teofiliny i zmniejszenie dawki teofiliny.Nie należy brać pod uwagę interakcji między produktem EXJADE a innymi substratami CYP1A2. wyłączony. Te same zalecenia dotyczące teofiliny dotyczą substancji metabolizowanych głównie przez cytochrom CYP1A2 o wąskim indeksie terapeutycznym (np. klozapina, tyzanidyna).
Jednoczesne podawanie produktu EXJADE i preparatów zobojętniających kwas zawierających glin nie było formalnie badane. Chociaż deferazyroks ma mniejsze powinowactwo do glinu niż do żelaza, nie zaleca się przyjmowania tabletek EXJADE z preparatami zobojętniającymi kwasy zawierającymi glin.
Jednoczesne podawanie preparatu EXJADE i witaminy C nie było formalnie badane. Dawki witaminy C do 200 mg dziennie nie wiązały się z niekorzystnymi konsekwencjami.
Nie zaobserwowano interakcji pomiędzy preparatem EXJADE i digoksyną u zdrowych dorosłych ochotników.
04.6 Ciąża i laktacja
Ciąża
Brak danych klinicznych dotyczących ekspozycji na deferazyroks w ciąży. Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję w dawkach uznanych za toksyczne dla matki (patrz punkt 5.3). Potencjalne ryzyko dla ludzi nie jest znane.
Jako środek ostrożności, nie zaleca się stosowania leku EXJADE w okresie ciąży, chyba że jest to absolutnie konieczne.
EXJADE może zmniejszać skuteczność hormonalnych środków antykoncepcyjnych (patrz punkt 4.5).
Czas karmienia
W badaniach na zwierzętach stwierdzono, że deferazyroks jest szybko i intensywnie wydzielany do mleka ludzkiego. Nie zaobserwowano wpływu na potomstwo. Nie wiadomo, czy deferazyroks przenika do mleka ludzkiego. Nie zaleca się karmienia piersią podczas przyjmowania leku EXJADE.
Płodność
Brak danych dotyczących płodności u ludzi.U zwierząt nie stwierdzono niekorzystnego wpływu na płodność samców ani samic (patrz punkt 5.3).
04.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Nie przeprowadzono badań dotyczących wpływu produktu EXJADE na zdolność prowadzenia pojazdów i obsługiwania maszyn Pacjenci, u których występują zawroty głowy, niezbyt częste działanie niepożądane, powinni zachować ostrożność podczas prowadzenia pojazdów lub obsługiwania maszyn (patrz punkt 4.8).
04.8 Działania niepożądane
Podsumowanie profilu bezpieczeństwa
Najczęstsze reakcje zgłaszane podczas przewlekłego leczenia produktem EXJADE u dorosłych i dzieci to zaburzenia żołądkowo-jelitowe u około 26% pacjentów (głównie nudności, wymioty, biegunka lub ból brzucha) oraz wysypka u około 7% pacjentów. Biegunkę zgłaszano najczęściej u dzieci w wieku od 2 do 5 lat oraz u osób w podeszłym wieku. Reakcje te są zależne od dawki, przeważnie o nasileniu łagodnym do umiarkowanego, zwykle przemijające i w większości przypadków ustępują nawet podczas kontynuowania leczenia.
Podczas badań klinicznych około 36% pacjentów doświadczyło wzrostu stężenia kreatyniny w surowicy > 33% w dwóch lub więcej kolejnych oznaczeniach, czasami powyżej górnej granicy normy. Były one zależne od dawki. Około dwie trzecie pacjentów, u których wystąpił wzrost stężenie kreatyniny w surowicy powróciło do poziomu poniżej 33% bez dostosowania dawki.U pozostałych pacjentów wzrost stężenia kreatyniny w surowicy nie zawsze reagował na zmniejszenie dawki lub przerwanie leczenia.W niektórych przypadkach po zmniejszeniu dawki obserwowano jedynie stabilizację stężenia kreatyniny w surowicy (patrz punkt 4.4).
W retrospektywnej metaanalizie 2102 dorosłych i pediatrycznych pacjentów z talasemią beta w trakcie transfuzji (w tym pacjentów o różnych cechach, takich jak intensywność transfuzji, dawka i czas trwania leczenia) leczonych w dwóch randomizowanych badaniach klinicznych i czterech otwartych badaniach trwających do pięciu lat zaobserwowano zmniejszenie średniego klirensu kreatyniny o 13,2% u pacjentów dorosłych (95% CI: -14,4%, -12,1%; n = 935) i 9, 9% u pacjentów pediatrycznych (95% CI: -11,1 %, -8,6%; n = 1142) w pierwszym roku leczenia. W podgrupie pacjentów obserwowanych przez ponad rok (n = 250 do pięciu lat) nie zaobserwowano dalszego spadku średniego poziomu klirensu kreatyniny w kolejnych latach.
W randomizowanym, podwójnie zaślepionym, kontrolowanym placebo, trwającym rok badaniu z udziałem pacjentów z niezależnymi od transfuzji zespołami talasemii i przeładowaniem żelazem, najczęstszymi zdarzeniami niepożądanymi związanymi z lekiem zgłaszanymi przez pacjentów leczonych produktem leczniczym EXJADE w dawce 10 mg/kg/kg mc. dnia biegunka (9,1%), wysypka (9,1%) i nudności (7,3%). Zmiany klirensu kreatyniny i kreatyniny w surowicy zgłaszano odpowiednio u 5,5% i 1,8% pacjentów leczonych produktem leczniczym EXJADE w dawce 10 mg/kg/dobę. pacjentów leczonych preparatem EXJADE 10 mg/kg/dobę.
Tabela działań niepożądanych
Działania niepożądane uszeregowano poniżej, stosując następującą konwencję: bardzo często (≥1 / 10); często (≥1/100,
Tabela 1
1 Działania niepożądane zgłaszane po wprowadzeniu produktu do obrotu Pochodzą one ze zgłoszeń spontanicznych, dla których nie zawsze jest możliwe bezpieczne ustalenie częstości lub związku przyczynowego z ekspozycją na lek.
Kamienie żółciowe i związane z nimi zaburzenia dróg żółciowych zgłaszano u około 2% pacjentów.Podwyższenie aktywności aminotransferaz zgłoszono jako działanie niepożądane leku u 2% pacjentów. Niezbyt często (0,3%) występowało zwiększenie aktywności aminotransferaz ponad 10-krotnie powyżej górnej granicy normy, wskazujące na zapalenie wątroby.Po wprowadzeniu produktu leczniczego do obrotu zgłaszano niewydolność wątroby, czasami prowadzącą do zgonu, zwłaszcza u pacjentów z istniejąca wcześniej marskość wątroby (patrz punkt 4.4). Po wprowadzeniu produktu do obrotu zgłaszano przypadki kwasicy metabolicznej. Większość z tych pacjentów miała zaburzenia czynności nerek, zaburzenia czynności kanalika nerkowego (zespół Fanconiego) lub biegunkę lub stany, w których zaburzenia równowagi kwasowo-zasadowej są znanym powikłaniem (patrz punkt 4.4). Ciężkie ostre zapalenie trzustki może potencjalnie występować jako powikłanie kamieni żółciowych (i powiązanych zaburzeń dróg żółciowych). Podobnie jak w przypadku innych terapii chelatujących żelazo, u pacjentów leczonych produktem leczniczym EXJADE niezbyt często obserwowano utratę słuchu o wysokiej częstotliwości i zmętnienie soczewki (wczesną zaćmę) (patrz punkt 4.4).
Populacja pediatryczna
Biegunkę zgłaszano częściej u dzieci w wieku od 2 do 5 lat niż u starszych pacjentów.
Tubulopatia nerkowa była zgłaszana głównie u dzieci i młodzieży z beta-talasemią leczonych produktem EXJADE.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie podejrzewanych działań niepożądanych występujących po dopuszczeniu produktu leczniczego do obrotu jest ważne, ponieważ umożliwia ciągłe monitorowanie stosunku korzyści do ryzyka produktu leczniczego. Osoby należące do fachowego personelu medycznego są proszone o zgłaszanie wszelkich podejrzewanych działań niepożądanych za pośrednictwem krajowego systemu zgłaszania. w „Załączniku V .
04.9 Przedawkowanie
Zgłaszano przypadki przedawkowania (2-3-krotność przepisanej dawki przez kilka tygodni). W jednym przypadku doprowadziło to do podklinicznego zapalenia wątroby, które ustąpiło po „przerwaniu leczenia. Pojedyncze dawki 80 mg/kg u przeładowanych żelazem pacjentów z talasemią powodowały łagodne nudności i biegunkę.
Ostre objawy przedawkowania mogą obejmować nudności, wymioty, ból głowy i biegunkę. Przedawkowanie można leczyć poprzez wywołanie wymiotów lub płukanie żołądka oraz leczenie objawowe.
05.0 WŁAŚCIWOŚCI FARMAKOLOGICZNE
05.1 Właściwości farmakodynamiczne
Grupa farmakoterapeutyczna: środek chelatujący żelazo.
Kod ATC: V03AC03.
Mechanizm akcji
Deferazyroks jest aktywnym chelatorem po podaniu doustnym, wysoce selektywnym w stosunku do żelaza (III). Jest to ligand trójkleszczowy, który wiąże żelazo z wysokim powinowactwem w stosunku 2:1. Deferazyroks wspomaga wydalanie żelaza, głównie z kałem.Deferazyroks ma małe powinowactwo do cynku i miedzi i nie powoduje stałego spadku stężenia tych metali w surowicy.
Efekty farmakodynamiczne
W badaniu metabolizmu równowagi żelaza u dorosłych pacjentów z talasemią z przeciążeniem żelazem, produkt EXJADE w dawkach dobowych 10, 20 i 40 mg/kg indukował średnie wydalanie netto odpowiednio 0,119, 0,329 i 0,445 mg Fe/kg masy ciała/dobę.
Skuteczność kliniczna i bezpieczeństwo
Preparat EXJADE badano u 411 dorosłych pacjentów (w wieku ≥16 lat) i 292 pacjentów pediatrycznych (w wieku 2 lat z niedokrwistością sierpowatokrwinkową i innymi niedokrwistościami wrodzonymi i nabytymi (zespoły mielodysplastyczne, zespół Diamonda-Blackfana, niedokrwistość aplastyczna i inne bardzo rzadkie niedokrwistości).
Codzienne leczenie dorosłych i dzieci, często poddawanych transfuzji, z talasemią beta w dawkach 20 i 30 mg/kg przez jeden rok, prowadziło do obniżenia wskaźników żelaza w całym organizmie; Stężenie żelaza w wątrobie zmniejszyło się średnio o około -0,4 i -8,9 mg Fe/g wątroby (sucha masa z biopsji), a ferrytyna w surowicy zmniejszyła się średnio o około -36 i -926 mcg/l. Przy tych samych dawkach stosunek wydalania żelaza do spożycia żelaza wynosił odpowiednio 1,02 (wskazując na bilans żelaza netto) i 1,67 (wskazując na „eliminację żelaza netto). dawki 10 mg/kg przez jeden rok mogą utrzymać poziom żelaza w wątrobie i ferrytyny w surowicy oraz indukować bilans żelaza netto u pacjentów otrzymujących rzadkie transfuzje lub erytrocytoaferezę.Ferrytyna w surowicy oceniana podczas comiesięcznego monitorowania odzwierciedlała zmiany stężenia żelaza w wątrobie, co wskazuje, że wzorzec ferrytyny w surowicy może być używany do monitorowania odpowiedzi na terapię. Ograniczone dane kliniczne (29 pacjentów z wyjściową prawidłową czynnością serca) z użyciem rezonansu magnetycznego wskazują, że leczenie preparatem EXJADE 10-30 mg/kg/dobę przez 1 rok może również obniżyć stężenie żelaza w sercu (średnio wzrost MRI T2* wynosił od 18,3 do 23,0 milisekund).
Główna analiza głównego badania porównawczego przeprowadzonego z udziałem 586 pacjentów z talasemią beta i obciążeniem żelazem w wyniku transfuzji krwi nie wykazała równoważności produktu EXJADE w stosunku do deferoksaminy w analizie całej populacji pacjentów. Analiza post-hoc tego badania wykazała, że w podgrupie pacjentów ze stężeniem żelaza w wątrobie ≥7 mg Fe/g suchej masy leczonych preparatem EXJADE (20 i 30 mg/kg) lub deferoksaminą (35 do ≥50 mg/kg), spełnione zostały kryteria równoważności. Jednak u pacjentów ze stężeniem żelaza w wątrobie
Badania przedkliniczne i kliniczne wykazały, że EXJADE może być aktywny jako deferoksamina, gdy jest stosowany w stosunku dawek 2:1 (np. dawka EXJADE jest liczbowo o połowę mniejsza od dawki deferoksaminy). Jednak to zalecenie dotyczące dawkowania nie zostało ocenione prospektywnie w badaniach klinicznych.
Dodatkowo u pacjentów ze stężeniem żelaza w wątrobie ≥7 mg Fe/g suchej masy, cierpiących na różne rzadkie anemie lub anemię sierpowatą, EXJADE do 20 i 30 mg/kg powoduje zmniejszenie stężenia żelaza w wątrobie i ferrytyny w surowicy porównywalne z uzyskanymi. u pacjentów z talasemią beta.
U pacjentów z zespołami talasemii niezależnymi od transfuzji krwi i obciążeniem żelazem leczenie produktem EXJADE oceniano w rocznym, randomizowanym, podwójnie zaślepionym badaniu kontrolowanym placebo. W badaniu porównywano skuteczność dwóch różnych schematów leczenia deferazyroksem (dawki początkowe 5 i 10 mg/kg/dobę, 55 pacjentów w każdym ramieniu) i odpowiadającego im placebo (56 pacjentów). 145 pacjentów dorosłych i 21 pacjentów pediatrycznych. zmiana stężenia żelaza w wątrobie (LIC) w stosunku do wartości wyjściowej po 12 miesiącach leczenia Jednym z drugorzędowych parametrów skuteczności była zmiana stężenia ferrytyny w surowicy między początkiem a czwartym trymetrem. u pacjentów leczonych preparatem EXJADE (dawka początkowa 10 mg/kg/dobę) i wzrosło o 0,38 mg Fe/g/sucha masa u pacjentów pacjenci leczeni placebo (p
Europejska Agencja Leków odroczyła obowiązek przedstawienia wyników badań produktu EXJADE w jednej lub kilku podgrupach populacji pediatrycznej w leczeniu przewlekłego obciążenia żelazem wymagającego terapii chelatacyjnej (informacje na temat stosowania u dzieci, patrz punkt 4.2).
05.2 Właściwości farmakokinetyczne
Wchłanianie
Deferazyroks wchłania się po podaniu doustnym, a mediana czasu do osiągnięcia maksymalnego stężenia w osoczu (Tmax) wynosi około 1,5 do 4 godzin. Całkowita biodostępność (AUC) deferazyroksu z tabletek EXJADE wynosi około 70% w porównaniu z dawką dożylną. Całkowita ekspozycja (AUC) była w przybliżeniu podwojona, gdy była przyjmowana z posiłkiem o wysokiej zawartości tłuszczu (zawartość tłuszczu > 50% kalorii) i wzrosła o około 50%, gdy była przyjmowana ze standardowym posiłkiem.Biodostępność (AUC) deferazyroksu była umiarkowana (około 13-25 %) wyższy, gdy jest przyjmowany 30 minut przed normalnymi lub wysokotłuszczowymi posiłkami.
Dystrybucja
Deferazyroks w wysokim stopniu (99%) wiąże się z białkami osocza, prawie wyłącznie z albuminą surowicy i ma małą objętość dystrybucji, około 14 litrów u dorosłych.
Biotransformacja
Glukuronidacja jest głównym szlakiem metabolicznym deferazyroksu, a następnie wydalanie z żółcią. Prawdopodobne jest wystąpienie dekoniugacji glukuronidów w jelicie, a następnie reabsorpcji (krążenie jelitowo-wątrobowe): W badaniu z udziałem zdrowych ochotników podanie cholestyraminy po podaniu pojedynczej dawki deferazyroksu powodowało 45% zmniejszenie ekspozycji na deferazyroks (AUC).
Deferazyroks jest głównie glukuronidowany przez UGT1A1 iw mniejszym stopniu UGT1A3. Katalizowany przez CYP450 (oksydacyjny) metabolizm deferazyroksu wydaje się być niewielki u ludzi (około 8%). In vitro nie zaobserwowano hamowania metabolizmu deferazyroksu przez hydroksymocznik.
Eliminacja
Deferazyroks i jego metabolity są wydalane głównie z kałem (84% dawki). Wydalanie nerkowe deferazyroksu i jego metabolitów jest minimalne (8% dawki), a średni okres półtrwania w fazie eliminacji (t1/2) wynosi od 8 do 16 godzin. Transportery MRP2 i MXR (BCRP) biorą udział w wydalaniu deferazyroksu z żółcią.
Liniowość / Nieliniowość
W warunkach stanu stacjonarnego Cmax i AUC0-24h deferazyroksu wzrastają w przybliżeniu liniowo wraz z dawką.W przypadku wielokrotnego dawkowania ekspozycja wzrasta o współczynnik akumulacji od 1,3 do 2,3.
Charakterystyka pacjentów
Pacjenci pediatryczni
Całkowita ekspozycja młodzieży (12 do ≤17 lat) i dzieci (2 do
Seks
Kobiety mają umiarkowanie niższy (17,5%) pozorny klirens deferazyroksu niż mężczyźni. Ponieważ dawkowanie jest dostosowywane indywidualnie w zależności od odpowiedzi, nie oczekuje się, że będzie to miało konsekwencje kliniczne.
Starsi pacjenci
Nie badano farmakokinetyki deferazyroksu u pacjentów w podeszłym wieku (w wieku 65 lat i starszych).
Niewydolność nerek lub wątroby
Nie badano farmakokinetyki deferazyroksu u pacjentów z niewydolnością nerek. Na farmakokinetykę deferazyroksu nie mają wpływu aktywności aminotransferaz wątrobowych do 5 razy większe od górnej granicy normy.
W badaniu klinicznym z zastosowaniem pojedynczej dawki 20 mg/kg deferazyroksu średnia ekspozycja wzrosła o 16% u osób z łagodnymi zaburzeniami czynności wątroby (klasa A w skali Childa-Pugha) i o 76% u osób z umiarkowanymi zaburzeniami czynności wątroby (klasa w skali Childa-Pugha). B) w porównaniu z osobami z prawidłową czynnością wątroby U osób z łagodnymi do umiarkowanych zaburzeniami czynności wątroby średnie Cmax deferazyroksu zwiększyło się o 22% U osób z ciężkimi zaburzeniami czynności wątroby (klasa C wg Childa-Pugha) ekspozycja wzrosła 2,8-krotnie (patrz punkty 4.2). oraz 4.4).
05.3 Przedkliniczne dane o bezpieczeństwie
Dane niekliniczne, oparte na konwencjonalnych badaniach dotyczących: farmakologia bezpieczeństwa, toksyczność po podaniu wielokrotnym, genotoksyczność lub potencjał rakotwórczy. Głównymi odkryciami były toksyczność nerkowa i zmętnienie soczewki (zaćma). Podobne dowody zaobserwowano u nowonarodzonych i młodocianych zwierząt. Uważa się, że toksyczność nerek jest głównie spowodowana utratą żelaza u zwierząt, które nie miały wcześniej przeładowania żelazem.
Testy genotoksyczności in vitro były negatywne (test Amesa, test aberracji chromosomowych) lub pozytywne (przesiewowe V79). Deferazyroks powodował powstawanie mikrojąder in vivo w szpiku kostnym, ale nie w wątrobie, u szczurów, którym nie podano ładunku żelaza w śmiertelnych dawkach. Nie zaobserwowano takich efektów u szczurów, którym nie podano ładunku żelaza. Deferazyroks nie wykazywał działania rakotwórczego po podaniu szczurom w 2-letnim badaniu i p53 +/- transgeniczne myszy heterozygotyczne w 6-miesięcznym badaniu.
Potencjał toksycznego wpływu na reprodukcję oceniano u szczurów i królików. Deferazyroks nie wykazywał działania teratogennego, ale powodował zwiększoną częstość zmian szkieletowych i martwych urodzeń u szczurów w dużych dawkach, które były silnie toksyczne dla matki nie przeładowanej żelazem. Deferazyroks nie miał żadnego innego wpływu na płodność lub reprodukcję.
06.0 INFORMACJE FARMACEUTYCZNE
06.1 Zaróbki
Monohydrat laktozy
Krospowidon typu A
Celuloza mikrokrystaliczna
Powidon
Laurylosiarczan sodu
Bezwodna krzemionka koloidalna
Stearynian magnezu
06.2 Niezgodność
Nie zaleca się dyspersji w napojach gazowanych lub mleku ze względu odpowiednio na pienienie i powolne dyspergowanie. Tabletek nie należy żuć ani połykać w całości.
06.3 Okres ważności
3 lata.
06.4 Specjalne środki ostrożności przy przechowywaniu
Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.
06.5 Rodzaj opakowania bezpośredniego i zawartość opakowania
Blistry PVC / PE / PVDC / Aluminium.
Opakowania zawierające 28, 84 lub 252 tabletki do sporządzania zawiesiny.
Nie wszystkie rozmiary opakowań mogą być wprowadzone na rynek.
06.6 Instrukcje użytkowania i obsługi
Brak specjalnych instrukcji.
07.0 PODMIOT POZWOLENIA NA DOPUSZCZENIE DO OBROTU
Novartis Europharm Limited
Frimley Business Park
Camberley GU16 7SR
Wielka Brytania
08.0 NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
UE / 1/06/356/001
037421017
UE / 1/06/356/002
037421029
UE / 1/06/356/007
09.0 DATA PIERWSZEGO ZEZWOLENIA LUB PRZEDŁUŻENIA ZEZWOLENIA
Data pierwszej autoryzacji: 28.08.2006
Data ostatniego przedłużenia: 28.08.2011
10.0 DATA ZMIAN TEKSTU
DCCE Lipiec 2015





-cos-cause-e-come-superarla.jpg)