Składniki aktywne: Sorafenib
Nexavar 200 mg tabletki powlekane
Dlaczego stosuje się Nexavar? Po co to jest?
Nexavar jest stosowany w leczeniu raka wątroby.
Nexavar jest również stosowany w leczeniu raka nerki (zaawansowany rak nerkowokomórkowy), gdy jest on w zaawansowanym stadium i gdy standardowe leczenie nie pomogło w jego zatrzymaniu lub jest uważane za nieodpowiednie.
Nexavar stosuje się w leczeniu raka tarczycy (zróżnicowanego raka tarczycy).
Nexavar jest tak zwanym inhibitorem wielu kinaz. Działa poprzez spowolnienie tempa wzrostu komórek rakowych i blokowanie dopływu krwi, który umożliwia wzrost komórek rakowych.
Przeciwwskazania Kiedy nie należy stosować produktu Nexavar
Nie przyjmować leku Nexavar
- jeśli pacjent ma uczulenie na sorafenib lub którykolwiek z pozostałych składników tego leku (wymienionych w punkcie 6).
Środki ostrożności dotyczące stosowania Informacje ważne przed przyjęciem leku Nexavar
Przed rozpoczęciem przyjmowania leku Nexavar należy omówić to z lekarzem lub farmaceutą.
Szczególnie uważaj na Nexavar
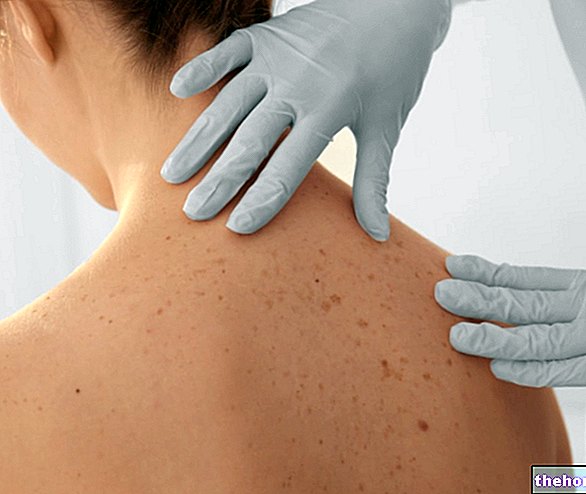
- Jeśli wystąpią problemy skórne. Nexavar może powodować wysypki i reakcje skórne, zwłaszcza na dłoniach i stopach. Skutki te zazwyczaj może leczyć lekarz. W przeciwnym razie lekarz może zawiesić leczenie lub całkowicie je przerwać.
- Jeśli masz wysokie ciśnienie krwi. Nexavar może powodować wzrost ciśnienia krwi; lekarz będzie regularnie kontrolował ciśnienie krwi i może przepisać leki na nadciśnienie.
- Jeśli masz problemy z krwawieniem lub jeśli przyjmujesz warfarynę lub fenprokomon. Leczenie lekiem Nexavar może prowadzić do zwiększonego ryzyka krwawienia. Jeśli pacjent przyjmuje warfarynę lub fenprokomon, leki rozrzedzające krew zapobiegające powstawaniu zakrzepów krwi, może wystąpić zwiększone ryzyko krwawienia.
- Jeśli masz bóle w klatce piersiowej lub problemy z sercem. Lekarz może podjąć decyzję o przerwaniu leczenia lub całkowitym jego zaprzestaniu.
- jeśli u pacjenta występuje choroba serca, taka jak „zaburzenie sygnału elektrycznego zwane” wydłużeniem odstępu QT ”.
- Jeśli masz mieć lub właśnie przeszłaś operację. Nexavar może wpływać na gojenie się ran. W przypadku planowanego zabiegu chirurgicznego leczenie lekiem Nexavar zostanie prawdopodobnie przerwane. Lekarz zdecyduje wtedy, kiedy go wycofać.
- Jeśli pacjent jest leczony irynotekanem lub docetakselem, które są również lekami przeciwnowotworowymi, Nexavar może nasilać działanie, a zwłaszcza działania niepożądane tych leków.
- Jeśli przyjmujesz neomycynę lub inne antybiotyki. Skuteczność leku Nexavar może być zmniejszona - jeśli u pacjenta występuje ciężka niewydolność wątroby, mogą wystąpić nasilone działania niepożądane podczas stosowania tego leku.
- jeśli u pacjenta występuje zmniejszona czynność nerek. Twój lekarz będzie monitorował równowagę wodno-elektrolitową.
- Płodność. Nexavar może zmniejszać płodność zarówno u mężczyzn, jak iu kobiet. Jeśli dotyczy to Ciebie, porozmawiaj ze swoim lekarzem.
- Podczas leczenia może wystąpić perforacja przewodu pokarmowego (patrz punkt 4: Możliwe działania niepożądane). W takim przypadku lekarz przerwie leczenie.
- Jeśli masz raka tarczycy, lekarz sprawdzi poziom wapnia i hormonu tarczycy we krwi.
Należy poinformować lekarza, jeśli którekolwiek z powyższych dotyczy Ciebie. Może być konieczne leczenie tych problemów, lekarz może zmienić dawkę leku Nexavar lub całkowicie przerwać leczenie (patrz również punkt 4: Możliwe działania niepożądane).
Dzieci i młodzież
Nexavar nie był jeszcze badany u dzieci i młodzieży.
Interakcje Jakie leki lub pokarmy mogą zmienić działanie leku Nexavar
Niektóre leki mogą wpływać na Nexavar lub być pod jego wpływem. Należy poinformować lekarza lub farmaceutę, jeśli pacjent przyjmuje, ostatnio przyjmował lub może przyjmować którykolwiek z leków z tej listy lub inne leki, w tym leki wydawane bez recepty:
- Ryfampicyna, neomycyna lub inne leki stosowane w leczeniu zakażeń (antybiotyki)
- Hypericum perforatum, znany również jako „dziurawiec zwyczajny, ziołowy lek na depresję
- Fenytoina, karbamazepina lub fenobarbital, leczenie padaczki i innych chorób
- Deksametazon, kortykosteroid stosowany w różnych chorobach
- Warfaryna lub fenprokomon, leki przeciwzakrzepowe stosowane w zapobieganiu zakrzepom krwi
- Doksorubicyna, kapecytabina, docetaksel, paklitaksel i irynotekan, stosowane w leczeniu nowotworów.
- Digoksyna, stosowana w leczeniu łagodnej lub umiarkowanej niewydolności serca
Ostrzeżenia Ważne jest, aby wiedzieć, że:
Ciąża i karmienie piersią
Należy unikać zajścia w ciążę podczas leczenia lekiem Nexavar. Jeśli pacjentka jest w wieku rozrodczym, podczas leczenia lekiem Nexavar musi stosować skuteczną antykoncepcję. Jeśli pacjentka zajdzie w ciążę podczas leczenia lekiem Nexavar, powinna natychmiast powiedzieć o tym lekarzowi, który zdecyduje, czy leczenie należy kontynuować, czy przerwać.
Nie należy karmić dziecka piersią podczas leczenia lekiem Nexavar, ponieważ lek ten może zaburzać wzrost i rozwój dziecka.
Prowadzenie i używanie maszyn
Nie ma powodu, aby sądzić, że Nexavar wpływa na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Dawka, sposób i czas podawania Jak stosować Nexavar: Dawkowanie
Zalecana dawka leku Nexavar dla dorosłych to dwie tabletki 200 mg dwa razy na dobę.
Odpowiadają one dziennej dawce 800 mg lub czterem tabletkom dziennie. Tabletki Nexavar należy przyjmować popijając szklanką wody, między posiłkami lub z pokarmami o niskiej lub średniej zawartości tłuszczu.Nie należy przyjmować tego leku z pokarmami bardzo tłustymi, ponieważ mogą one zmniejszać ich skuteczność. Jeśli planujesz spożywać bardzo tłuste potrawy, zażywaj tabletki co najmniej 1 godzinę przed lub 2 godziny po obiedzie. Lek należy zawsze stosować zgodnie z zaleceniami lekarza. Jeśli nie masz pewności, skonsultuj się z lekarzem lub farmaceutą.
Ważne jest, aby przyjmować ten lek codziennie o tej samej porze, aby utrzymać stałe stężenie we krwi.
Ten lek jest zwykle przyjmowany tak długo, jak zauważone są korzyści kliniczne i nie ma niemożliwych do zniesienia skutków ubocznych.
Przedawkowanie Co zrobić, jeśli pacjent przyjął zbyt dużą dawkę leku Nexavar
Przyjęcie większej niż zalecana dawki leku Nexavar
Należy natychmiast poinformować lekarza, jeśli pacjent lub ktokolwiek inny przyjął dawkę większą niż przepisana. Zażycie zbyt dużej ilości leku Nexavar zwiększa prawdopodobieństwo wystąpienia lub poważniejszych działań niepożądanych, zwłaszcza biegunki i reakcji skórnych. Lekarz może zalecić przerwanie przyjmowania tego leku.
Pominięcie przyjęcia leku Nexavar
Jeśli zapomniałeś przyjąć dawkę, zażyj ją tak szybko, jak sobie przypomnisz. Jeśli kolejna dawka jest wkrótce po, zapomnij o pominiętej dawce i kontynuuj z freq
Skutki uboczne Jakie są skutki uboczne Nexavar
Jak każdy lek, lek ten może powodować działania niepożądane, chociaż nie u każdego one wystąpią. Ten lek może również zmieniać wyniki niektórych badań krwi.
Bardzo częste:
może dotyczyć więcej niż 1 na 10 osób
- biegunka
- złe samopoczucie (nudności)
- uczucie osłabienia lub zmęczenia (zmęczenie)
- ból (w tym ból jamy ustnej, brzucha, ból głowy, ból kości, ból nowotworowy)
- wypadanie włosów (łysienie)
- zaczerwienienie lub ból dłoni lub podeszew stóp (reakcja skórna ręka-stopa)
- swędzenie lub wysypka
- On wymiotował
- krwawienie (w tym krwotok w mózgu, ścianie jelita i drogach oddechowych)
- wysokie ciśnienie krwi lub podwyższone ciśnienie krwi (nadciśnienie)
- infekcje
- utrata apetytu (anoreksja)
- zaparcie
- ból stawów (artralgia)
- gorączka
- utrata wagi
- suchość skóry
Pospolity:
może dotyczyć nie więcej niż 1 na 10 osób
- choroba grypopodobna
- niestrawność (niestrawność)
- trudności w połykaniu (dysfagia)
- zapalenie lub suchość jamy ustnej, ból języka (zapalenie jamy ustnej i zapalenie błony śluzowej)
- niski poziom wapnia we krwi (hipokalcemia)
- małe stężenie potasu we krwi (hipokaliemia)
- ból mięśni (mialgia)
- zaburzenia wrażliwości palców rąk i nóg, w tym mrowienie i drętwienie (obwodowa neuropatia czuciowa)
- depresja
- problemy z erekcją (impotencja)
- zmiany głosu (dysfonia)
- trądzik
- stan zapalny, sucha lub łuszcząca się skóra (zapalenie skóry, łuszczenie się skóry)
- niewydolność serca
- zawał serca (zawał mięśnia sercowego) lub ból w klatce piersiowej
- szum w uszach (dzwonienie w uszach)
- niewydolność nerek
- wysoki poziom białka w moczu (białkomocz)
- ogólne osłabienie lub utrata siły (astenia)
- zmniejszona liczba białych krwinek (leukopenia i neutropenia)
- zmniejszona liczba czerwonych krwinek (niedokrwistość)
- mała liczba płytek krwi (trombocytopenia)
- zapalenie mieszków włosowych (zapalenie mieszków włosowych)
- zmniejszona aktywność tarczycy (niedoczynność tarczycy)
- niski poziom sodu we krwi (hiponatremia)
- zmiany w poczuciu smaku (dysgeusia)
- zaczerwienienie twarzy i często innych obszarów skóry (zaczerwienienie)
- katar (katar)
- zgaga (choroba refluksowa przełyku)
- rak skóry (rogowiak kolczystokomórkowy / rak kolczystokomórkowy skóry)
- pogrubienie zewnętrznej warstwy skóry (hiperkeratoza)
- nagły mimowolny skurcz mięśnia (skurcze mięśni)
Niezwykły:
może dotyczyć nie więcej niż 1 na 100 osób
- zapalenie żołądka (zapalenie żołądka)
- ból brzucha (brzucha) z powodu zapalenia trzustki, zapalenia pęcherzyka żółciowego i / lub dróg żółciowych
- zażółcenie skóry lub oczu (żółtaczka) spowodowane wysokim poziomem barwników żółciowych (hiperbilirubinemia)
- reakcje typu alergicznego (w tym reakcje skórne i pokrzywka)
- odwodnienie
- powiększenie piersi (ginekomastia)
- trudności w oddychaniu (choroba płuc)
- wyprysk
- nadmierna aktywność tarczycy (nadczynność tarczycy)
- liczne wysypki skórne (rumień wielopostaciowy)
- wysokie ciśnienie krwi
- perforacja przewodu pokarmowego
- odwracalny obrzęk w tylnej części mózgu, który może być związany z bólem głowy, zaburzeniami świadomości, drgawkami i objawami wizualnymi, w tym utratą wzroku (odwracalna tylna leukoencefalopatia)
- nagła, ciężka reakcja alergiczna (reakcja anafilaktyczna)
Rzadki:
może dotyczyć nie więcej niż 1 na 1000 osób
- reakcja alergiczna z obrzękiem skóry (np. twarzy, języka), który może powodować trudności w oddychaniu i połykaniu (obrzęk naczynioruchowy)
- nieprawidłowy rytm serca (wydłużenie odstępu QT)
- Zapalenie wątroby, które może prowadzić do nudności, wymiotów, bólu brzucha i żółtaczki (polekowe zapalenie wątroby)
- „wysypka podobna do oparzeń słonecznych na skórze uprzednio wystawionej na radioterapię i może być ciężka (popromienne zapalenie skóry)
- ciężkie reakcje skórne i (lub) błon śluzowych, które mogą obejmować bolesne pęcherze i gorączkę, z odwarstwieniem dużych obszarów skóry (zespół Stevensa-Johnsona i toksyczna nekroliza naskórka)
- nieprawidłowy rozpad mięśni, który może prowadzić do problemów z nerkami (rabdomioliza)
- uszkodzenie nerek, które powoduje utratę dużej ilości białka z moczem (zespół nerczycowy)
- zapalenie naczyń krwionośnych skóry, które może objawiać się wysypką (leukocytoklastyczne zapalenie naczyń)
Nieznany:
częstotliwości nie można oszacować na podstawie dostępnych danych
- zaburzenia czynności mózgu, które mogą być związane np. z sennością, zmianami behawioralnymi lub splątaniem (encefalopatia)
Zgłaszanie skutków ubocznych
Jeśli wystąpią jakiekolwiek objawy niepożądane, w tym wszelkie możliwe działania niepożądane niewymienione w tej ulotce, należy porozmawiać z lekarzem lub farmaceutą. Działania niepożądane można również zgłaszać bezpośrednio za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V. Zgłaszanie działań niepożądanych może pomóc w uzyskaniu większej ilości informacji na temat bezpieczeństwa tego leku.
Wygaśnięcie i przechowywanie
Lek należy przechowywać w miejscu niewidocznym i niedostępnym dla dzieci.
Nie stosować tego leku po upływie terminu ważności zamieszczonego na pudełku po EXP i na każdym blistrze po EXP. Termin ważności odnosi się do ostatniego dnia tego miesiąca.
Nie przechowywać tego leku w temperaturze powyżej 25°C.
Nie należy wyrzucać żadnych leków do kanalizacji ani domowych pojemników na odpadki. Należy zapytać farmaceutę, jak usunąć leki, których się już nie używa.Pomoże to chronić środowisko.
Co zawiera lek Nexavar
- Substancją czynną jest sorafenib. Każda tabletka powlekana zawiera 200 mg sorafenibu (w postaci tosylanu).
- Pozostałe składniki to: Rdzeń tabletki: kroskarmeloza sodowa, celuloza mikrokrystaliczna, hypromeloza, laurylosiarczan sodu i stearynian magnezu. Otoczka tabletki: hypromeloza, makrogol, dwutlenek tytanu (E 171) i czerwony tlenek żelaza (E 172)
Jak wygląda lek Nexavar i co zawiera opakowanie
Tabletki powlekane Nexavar 200 mg są czerwone i okrągłe, z krzyżem Bayer po jednej stronie i liczbą „200” po drugiej.Są one dostarczane w pudełkach tekturowych po 112 tabletek, zawierających cztery przezroczyste blistry kalendarzowe po 28 tabletek każdy.
Ulotka pakietu źródłowego: AIFA (Włoska Agencja Leków). Treść opublikowana w styczniu 2016 r. Przedstawione informacje mogą być nieaktualne.
Aby mieć dostęp do najbardziej aktualnej wersji, warto wejść na stronę AIFA (Włoskiej Agencji Leków). Zastrzeżenie i przydatne informacje.
01.0 NAZWA PRODUKTU LECZNICZEGO
NEXAVAR 200 MG
02.0 SKŁAD JAKOŚCIOWY I ILOŚCIOWY
Każda tabletka powlekana zawiera 200 mg sorafenibu (w postaci tosylanu).
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
03.0 POSTAĆ FARMACEUTYCZNA
Tabletka powlekana (tabletka).
Czerwone, okrągłe, obustronnie wypukłe tabletki powlekane oznaczone krzyżykiem Bayera po jednej stronie i liczbą „200” po drugiej.
04.0 INFORMACJE KLINICZNE
04.1 Wskazania terapeutyczne
rak wątroby
Nexavar jest wskazany w leczeniu raka wątrobowokomórkowego (patrz punkt 5.1).
Rak nerkowokomórkowy
Nexavar jest wskazany do leczenia pacjentów z zaawansowanym rakiem nerkowokomórkowym, u których wcześniejsza terapia interferonem alfa lub interleukiną-2 okazała się nieskuteczna, lub którzy zostali uznani za niekwalifikujących się do takiej terapii.
Zróżnicowany rak tarczycy
Nexavar jest wskazany w leczeniu pacjentów z miejscowo zaawansowanym lub przerzutowym, postępującym, opornym na radiojody zróżnicowanym rakiem tarczycy (brodawkowatym / pęcherzykowym / komórka Hurthle'a).
04.2 Dawkowanie i sposób podawania
Leczenie preparatem Nexavar powinno odbywać się pod nadzorem lekarza mającego doświadczenie w stosowaniu terapii przeciwnowotworowych.
Dawkowanie
Zalecana dawka produktu Nexavar dla dorosłych to 400 mg sorafenibu (dwie tabletki 200 mg) dwa razy na dobę (co odpowiada całkowitej dawce dobowej 800 mg).
Leczenie należy kontynuować tak długo, jak obserwuje się korzyści kliniczne lub do momentu pojawienia się niedopuszczalnych działań toksycznych.
Dostosowanie dozowania
Postępowanie w przypadku podejrzewanych działań niepożądanych leku może wymagać czasowego przerwania lub zmniejszenia dawki sorafenibu.
Gdy konieczne jest zmniejszenie dawki podczas leczenia raka wątrobowokomórkowego (rak wątrobowokomórkowy, HCC) i raka nerkowokomórkowego (rak nerkowokomórkowyRCC), dawkę produktu Nexavar należy zmniejszyć do dwóch tabletek 200 mg sorafenibu raz na dobę (patrz punkt 4.4).
Gdy konieczne jest zmniejszenie dawki podczas leczenia zróżnicowanego raka tarczycy (zróżnicowany rak tarczycyDTC), dawkę produktu Nexavar należy zmniejszyć do 600 mg sorafenibu na dobę w dawkach podzielonych (dwie tabletki 200 mg i jedna tabletka 200 mg w odstępie 12 godzin).
Jeśli konieczne jest dalsze zmniejszenie dawki, Nexavar można zmniejszyć do 400 mg sorafenibu na dobę w dawkach podzielonych (dwie tabletki 200 mg w odstępie 12 godzin) i, w razie potrzeby, dodatkowo zmniejszyć do jednej tabletki 200 mg raz na dobę. działania niepożądane, dawkę produktu Nexavar można zwiększyć.
Populacja pediatryczna
Bezpieczeństwo i skuteczność produktu Nexavar u starszych dzieci i młodzieży
Populacja osób starszych
W przypadku osób w podeszłym wieku (pacjenci w wieku powyżej 65 lat) nie jest konieczne dostosowanie dawki.
Zaburzenia czynności nerek
Nie ma konieczności dostosowania dawki u pacjentów z łagodnymi, umiarkowanymi lub ciężkimi zaburzeniami czynności nerek. Brak danych dotyczących pacjentów dializowanych (patrz punkt 5.2).
U pacjentów z ryzykiem niewydolności nerek wskazane jest monitorowanie równowagi wodno-elektrolitowej.
Zaburzenia czynności wątroby
Nie ma konieczności dostosowania dawki u pacjentów z zaburzeniami czynności wątroby A lub B w skali Child Pugh (od łagodnego do umiarkowanego). Brak danych dotyczących pacjentów z ciężkimi zaburzeniami czynności wątroby w skali Child Pugh C (patrz punkty 4.4 i 5.2).
Sposób podawania
Do stosowania doustnego
Sorafenib należy podawać między posiłkami lub z posiłkiem o niskiej lub umiarkowanej zawartości tłuszczu. Jeśli pacjent zamierza spożyć posiłek o dużej zawartości tłuszczu, tabletki sorafenibu należy przyjmować co najmniej godzinę przed posiłkiem lub dwie godziny po posiłku. Tabletki należy połykać, popijając szklanką wody.
04.3 Przeciwwskazania
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
04.4 Specjalne ostrzeżenia i odpowiednie środki ostrożności dotyczące stosowania
Toksyczność dermatologiczna
Reakcja skórna ręka-stopa (erytrodyzestezja dłoniowo-podeszwowa) e wysypka reprezentują najczęstsze działania niepożądane sorafenibu. Wysypka a reakcja skórna dłoni i stóp jest zwykle stopnia 1 i 2, zgodnie z i Wspólne kryteria toksyczności (CTC) i zwykle pojawiają się w ciągu pierwszych sześciu tygodni leczenia sorafenibem. Postępowanie w przypadku toksyczności dermatologicznej może obejmować terapie miejscowe w celu złagodzenia objawów, czasowe przerwanie leczenia i (lub) zmianę dawki sorafenibu lub, w ciężkich lub utrzymujących się przypadkach, definitywne przerwanie jego podawania (patrz punkt 4.8).
Nadciśnienie
Większą częstość występowania nadciśnienia tętniczego obserwowano u pacjentów leczonych sorafenibem.U tych pacjentów nadciśnienie było zwykle łagodne do umiarkowanego, występowało we wczesnych stadiach leczenia i reagowało na standardowe leczenie przeciwnadciśnieniowe. Ciśnienie krwi powinno być regularnie monitorowane i leczone w razie potrzeby zgodnie z aktualną praktyką medyczną. W przypadku ciężkiego lub utrzymującego się nadciśnienia lub przełomu nadciśnieniowego, pomimo rozpoczęcia leczenia przeciwnadciśnieniowego, zaleca się rozważenie całkowitego przerwania podawania sorafenibu (patrz punkt 4.8).
Krwotok
Ryzyko krwawienia może wzrosnąć po podaniu sorafenibu. Jeśli epizod krwawienia wymaga interwencji medycznej, zaleca się rozważenie całkowitego przerwania podawania sorafenibu (patrz punkt 4.8).
Niedokrwienie serca i/lub zawał serca
W randomizowanym, kontrolowanym placebo badaniu z podwójnie ślepą próbą (badanie 1, patrz punkt 5.1), częstość występowania zawału serca lub niedokrwienia rozpoczynającego się od leczenia była większa w grupie sorafenibu (4,9%) niż w grupie otrzymującej placebo ( W badaniu 3 (patrz punkt 5.1) częstość występowania zawału serca lub niedokrwienia serca w trakcie leczenia wynosiła 2,7% u pacjentów leczonych sorafenibem i 1,3% u pacjentów otrzymujących placebo. Z badań tych wykluczono pacjentów z niestabilną chorobą wieńcową lub niedawno przebytym zawałem mięśnia sercowego. U pacjentów, u których rozwinie się niedokrwienie i (lub) zawał serca, należy rozważyć konieczność czasowego lub stałego przerwania leczenia sorafenibem (patrz punkt 4.8).
Wydłużenie odstępu QT
Wykazano, że sorafenib wydłuża odstęp QT/QTc (patrz punkt 5.1), co może prowadzić do zwiększonego ryzyka arytmii komorowej.Storafenib należy stosować ostrożnie u pacjentów, u których występuje wydłużenie odstępu QTc lub może dojść do jego wydłużenia, np. u pacjentów z wrodzonym wydłużonym odstępem QT Zespół, pacjenci leczeni dużą skumulowaną dawką antracyklin, pacjenci przyjmujący niektóre leki antyarytmiczne lub inne leki, które mogą prowadzić do wydłużenia odstępu QT oraz pacjenci z zaburzeniami elektrolitowymi, np. hipokaliemia, hipokalcemia lub hipomagnezemia. oraz pomiary elektrolitów (magnez, potas i wapń) powinny być wykonywane w okresie leczenia.
Perforacja przewodu pokarmowego
Perforacja przewodu pokarmowego jest rzadkim zdarzeniem i została zgłoszona u mniej niż 1% pacjentów przyjmujących sorafenib. W niektórych przypadkach nie było związku z wyraźnym guzem w jamie brzusznej. W przypadku perforacji przewodu pokarmowego należy przerwać podawanie sorafenibu (patrz punkt 4.8).
Zaburzenia czynności wątroby
Brak danych dotyczących pacjentów z ciężkimi zaburzeniami czynności wątroby (stopień C w skali Child-Pugh). U takich pacjentów ekspozycja może być zwiększona, ponieważ sorafenib jest eliminowany głównie przez wątrobę (patrz punkty 4.2 i 5.2).
Jednoczesne podawanie warfaryny
Rzadkie epizody krwawienia lub wzrost INR (Międzynarodowy Znormalizowany
Stosunek) u niektórych pacjentów przyjmujących warfarynę podczas leczenia sorafenibem. Pacjentów leczonych warfaryną lub fenprokumonem należy regularnie monitorować pod kątem zmian czasu protrombinowego, INR lub klinicznie istotnych epizodów krwawienia (patrz punkty 4.5 i 4.8).
Powikłania w gojeniu się ran
Nie przeprowadzono formalnych badań nad wpływem sorafenibu na gojenie się ran. Ze względów ostrożności u pacjentów poddawanych dużym zabiegom chirurgicznym zaleca się czasowe zawieszenie leczenia sorafenibem. Doświadczenie kliniczne dotyczące tego, kiedy należy wznowić leczenie po dużym zabiegu chirurgicznym, jest ograniczone. Dlatego decyzja o wznowieniu leczenia sorafenibem po dużym zabiegu chirurgicznym powinna opierać się na klinicznej ocenie odpowiedniego gojenia się rany.
Populacja osób starszych
Zgłaszano przypadki niewydolności nerek. Dlatego należy rozważyć monitorowanie czynności nerek.
Interakcje między lekami
Zaleca się ostrożność podczas podawania sorafenibu z substancjami, które są metabolizowane i (lub) eliminowane głównie przez szlak UGT1A1 (np. irynotekan) lub UGT1A9 (patrz punkt 4.5).
Zaleca się zachowanie ostrożności w przypadku jednoczesnego podawania sorafenibu i docetakselu (patrz punkt 4.5).
Skojarzenie z neomycyną lub innymi antybiotykami mogącymi powodować poważne zaburzenia ekologiczne mikroflory przewodu pokarmowego może prowadzić do zmniejszenia biodostępności sorafenibu (patrz punkt 4.5).Przed rozpoczęciem leczenia należy ocenić ryzyko zmniejszenia stężenia sorafenibu w osoczu. przebieg leczenia antybiotykami.
Wyższą śmiertelność obserwowano u pacjentów z płaskonabłonkowym rakiem płuca leczonych sorafenibem w skojarzeniu z chemioterapią opartą na platynie.
W dwóch randomizowanych badaniach klinicznych, w których badano pacjentów z niedrobnokomórkowym rakiem płuca (Niedrobnokomórkowego raka płuca, NSCLC), współczynnik ryzyka (HR) dla przeżycia całkowitego w podgrupie pacjentów z płaskonabłonkowym rakiem płuca wyniósł 1,81 (95% CI 1,19, 2,74) u pacjentów leczonych sorafenibem w połączeniu z paklitakselem/karboplatyną i 1,22 (95% CI 0,82; 1,80) u pacjentów leczonych sorafenibem dodatkowo do terapii gemcytabiną/cisplatyną. Nie zaobserwowano głównej przyczyny zgonu, ale zaobserwowano zwiększoną częstość występowania niewydolności oddechowej, krwawień i infekcji u pacjentów leczonych sorafenibem w połączeniu z terapią opartą na platynie.
Ostrzeżenia dotyczące patologii
Zróżnicowany rak tarczycy (DTC)
Przed rozpoczęciem leczenia zaleca się, aby lekarze dokładnie ocenili indywidualne rokowanie pacjenta na podstawie maksymalnej wielkości zmiany (patrz punkt 5.1), objawów związanych z chorobą (patrz punkt 5.1) i tempa progresji.
Postępowanie w przypadku podejrzewanych działań niepożądanych leku może wymagać „czasowego przerwania lub zmniejszenia dawki sorafenibu. W badaniu 5 (patrz punkt 5.1) 37% pacjentów czasowo przerwało leczenie, a 35% zmniejszyło dawkę już w 1. cyklu leczenia sorafenibem.
Zmniejszenie dawki było tylko częściowo skuteczne w łagodzeniu działań niepożądanych, dlatego zaleca się wielokrotną ocenę korzyści i ryzyka, biorąc pod uwagę aktywność przeciwnowotworową i tolerancję.
Krwotok w DTC
Ze względu na potencjalne ryzyko krwotoku nacieki tchawicy, oskrzeli i przełyku muszą być leczone miejscowo przed podaniem sorafenibu pacjentom z DTC.
Hipokalcemia w DTC
Podczas stosowania sorafenibu u pacjentów z DTC zaleca się ścisłe monitorowanie stężenia wapnia we krwi. W badaniach klinicznych hipokalcemia była częstsza i cięższa u pacjentów z DTC, szczególnie u pacjentów z niedoczynnością przytarczyc w wywiadzie, w porównaniu z pacjentami z rakiem nerkowokomórkowym lub rakiem wątroby. pacjenci z DTC leczeni sorafenibem (patrz punkt 4.8). Ciężką hipokalcemię należy skorygować, aby zapobiec powikłaniom, takim jak wydłużenie odstępu QT lub zaburzenia typu torsade de pointes (patrz punkt dotyczący wydłużenia odstępu QT).
Tłumienie TSH w DTC
W badaniu 5 (patrz punkt 5.1) u pacjentów leczonych sorafenibem obserwowano zwiększenie stężenia TSH o ponad 0,5 mU/l. Zaleca się ścisłe monitorowanie poziomu TSH podczas stosowania sorafenibu u pacjentów z DTC.
Rak nerkowokomórkowy
Pacjenci wysokiego ryzyka, zgodnie z definicją grupy prognostycznej MSKCC (Memorial Sloan Kettering Cancer Center), nie zostały uwzględnione w badaniu klinicznym III fazy dotyczącym raka nerkowokomórkowego (patrz badanie 1 w punkcie 5.1), a stosunek korzyści do ryzyka u tych pacjentów nie został ustalony.
04.5 Interakcje z innymi produktami leczniczymi i inne formy interakcji
Induktory enzymów metabolicznych
Podawanie ryfampicyny przez 5 dni przed podaniem pojedynczej dawki sorafenibu powodowało średnie zmniejszenie AUC sorafenibu o 37% Inne induktory CYP3A4 i/lub glukuronidacji (np. dziurawiec dziurawy znany również jako „ziele dziurawca”, fenytoina, karbamazepina, fenobarbital i deksametazon) mogą zwiększać metabolizm sorafenibu, a tym samym zmniejszać jego stężenie.
Inhibitory CYP3A4
Ketokonazol, silny inhibitor CYP3A4, podawany raz na dobę przez 7 dni zdrowym ochotnikom płci męskiej nie zmieniał średniej wartości AUC pojedynczej dawki 50 mg sorafenibu.Te dane sugerują, że kliniczne interakcje farmakokinetyczne sorafenibu z inhibitorami CYP3A4 są mało prawdopodobne.
Substraty CYP2B6, CYP2C8 i CYP2C9
In vitro sorafenib hamuje CYP2B6, CYP2C8 i CYP2C9 z prawie równą siłą. Jednak w klinicznych badaniach farmakokinetycznych jednoczesne podawanie sorafenibu 400 mg dwa razy na dobę z cyklofosfamidem, będącym substratem CYP2B6 lub paklitakselem, substratem CYP2C8, nie powodowało klinicznie istotnego hamowania.Te dane sugerują, że sorafenib w zalecanej dawce 400 mg dwa razy dziennie, może nie być inhibitorem in vivo CYP2B6 lub CYP2C8.
Ponadto jednoczesne leczenie sorafenibem i warfaryną, substratem CYP2C9, nie prowadziło do zmian średniego PT-INR w porównaniu z placebo. Dlatego też ryzyko „zahamowania” in vivo istotny klinicznie CYP2C9 przez sorafenib można uznać za niski. Jednak u pacjentów przyjmujących warfarynę lub fenprokumon należy regularnie monitorować INR (patrz punkt 4.4).
Substraty CYP3A4, CYP2D6 i CYP2C19
Jednoczesne podawanie sorafenibu i midazolamu, dekstrometorfanu lub omeprazolu, które są substratami dla cytochromów odpowiednio CYP3A4, CYP2D6 i CYP2C19, nie zmieniało ekspozycji na te leki, co wskazuje, że sorafenib nie jest ani inhibitorem, ani induktorem tych izoenzymów. , kliniczne interakcje farmakokinetyczne sorafenibu z substratami tych enzymów są mało prawdopodobne.
Substraty UGT1A1 i UGT1A9
In vitro, sorafenib hamował glukuronidację przez UGT1A1 i UGT1A9. Kliniczne znaczenie tego odkrycia nie jest znane (patrz poniżej i punkt 4.4).
Edukacja in vitro na indukcję enzymów układu CYP
Aktywność CYP1A2 i CYP3A4 nie uległa zmianie po ekspozycji hodowli ludzkich hepatocytów na sorafenib, co wskazuje, że jest mało prawdopodobne, aby sorafenib był induktorem CYP1A2 i CYP3A4.
Substraty do P-gp
In vitroWykazano, że sorafenib hamuje białko transportowe p-glikoproteinę (P-gp). W przypadku jednoczesnego leczenia sorafenibem nie można wykluczyć wzrostu stężenia substratów P-gp w osoczu, takich jak digoksyna.
Związek z innymi lekami przeciwnowotworowymi
W badaniach klinicznych sorafenib podawano z wieloma innymi lekami przeciwnowotworowymi w ich powszechnie stosowanych dawkach, w tym gemcytabiną, cisplatyną, oksaliplatyną, paklitakselem, karboplatyną, kapecytabiną, doksorubicyną, irynotekanem, docetakselem i cyklofosfamidem. Sorafenib nie miał klinicznie istotnego wpływu na farmakokinetykę gemcytabiny, cisplatyny, karboplatyny, oksaliplatyny ani cyklofosfamidu.
Paklitaksel / karboplatyna
• Podawanie paklitakselu (225 mg/m2) i karboplatyny (AUC = 6) z sorafenibem (≤ 400 mg dwa razy dziennie), z 3-dniową przerwą w podawaniu sorafenibu (poprzednie dwa dni i dzień podawania paklitakselu/karboplatyny ), nie miał znaczącego wpływu na farmakokinetykę paklitakselu.
• Jednoczesne podawanie paklitakselu (225 mg/m2, raz na 3 tygodnie) i karboplatyny (AUC = 6) z sorafenibem (400 mg dwa razy na dobę, bez przerwy w podawaniu sorafenibu) spowodowało 47% wzrost ekspozycji na sorafenib, 29% Zwiększenie ekspozycji na paklitaksel i 50% wzrost ekspozycji na paklitaksel 6-OH Nie miało to wpływu na farmakokinetykę karboplatyny.
Dane te wskazują, że nie ma konieczności dostosowania dawkowania w przypadku jednoczesnego podawania paklitakselu i karboplatyny z sorafenibem z 3-dniową przerwą w podawaniu sorafenibu (dwa dni przed i w dniu podania paklitakselu/karboplatyny). zwiększona ekspozycja na sorafenib i paklitaksel wkrótce po jednoczesnym podaniu sorafenibu bez przerwy w dawkowaniu.
Kapecytabina
Jednoczesne podawanie kapecytabiny (750-1050 mg/m2 dwa razy na dobę, dni 1-14 co 21 dni) i sorafenibu (200 lub 400 mg dwa razy na dobę bez przerwy w dawkowaniu) nie powodowało istotnych zmian w ekspozycji na sorafenib, ale 15 -50% wzrost ekspozycji na kapecytabinę i 0-52% wzrost ekspozycji na 5-FU.Kliniczne znaczenie tych niewielkich, umiarkowanych wzrostów ekspozycji na 5-FU nie jest znane, kapecytabina i 5-FU podczas jednoczesnego podawania z sorafenibem.
Doksorubicyna / Irynotekan
Jednoczesne leczenie sorafenibem powodowało zwiększenie AUC doksorubicyny o 21%.W przypadku podawania z irynotekanem, którego metabolit SN-38 jest następnie metabolizowany przez szlak UGT1A1, zaobserwowano 67-120% wzrost „AUC SN-38 i 26 - 42% w AUC irynotekanu." Znaczenie kliniczne tych danych nie jest znane (patrz punkt 4.4).
Docetaksel
Docetaksel (dawka 75 lub 100 mg/m2 co 21 dni) podawany jednocześnie z sorafenibem (200 mg lub 400 mg dwa razy na dobę w dniach 2 do 19 21-dniowego cyklu leczenia, przy czym „ 3 dni odpowiadają podawaniu docetakselu ) powodowało zwiększenie AUC i Cmax docetakselu odpowiednio o 36-80% i 16-32%.Należy zachować ostrożność podczas równoczesnego podawania z sorafenibem i docetakselem (patrz punkt 4.4).
Stowarzyszenie z innymi agentami
Neomycyna
Połączenie z neomycyną, nieukładowym środkiem przeciwdrobnoustrojowym stosowanym do eradykacji flory żołądkowo-jelitowej, zaburza recyrkulację jelitowo-wątrobową sorafenibu (patrz punkt 5.2, Biotransformacja i metabolizm), powodując zmniejszenie ekspozycji na sorafenib. dni średnia ekspozycja na sorafenib zmniejszyła się o 54%. Skutki innych antybiotyków nie zostały zbadane, ale najprawdopodobniej będą zależeć od ich zdolności do zakłócania drobnoustrojów o aktywności glukuronidazy.
04.6 Ciąża i laktacja
Ciąża
Brak danych dotyczących stosowania sorafenibu u kobiet w ciąży Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję, w tym wady rozwojowe (patrz punkt 5.3). Wykazano, że sorafenib i jego metabolity przenikają przez łożysko u szczurów i oczekuje się, że sorafenib ma szkodliwe działanie Sorafenibu nie należy stosować w okresie ciąży, chyba że jest to bezwzględnie konieczne i tylko po dokładnym rozważeniu potrzeb matki i ryzyka dla płodu.
Kobiety w wieku rozrodczym powinny podczas leczenia stosować skuteczną antykoncepcję.
Czas karmienia
Nie wiadomo, czy sorafenib przenika do mleka ludzkiego. U zwierząt sorafenib i (lub) jego metabolity przenikają do mleka.Ponieważ sorafenib może zaburzać wzrost i rozwój noworodka (patrz punkt 5.3), kobiety powinny przerwać karmienie piersią podczas leczenia sorafenibem.
Płodność
Badania na zwierzętach wykazują, że sorafenib może zaburzać płodność samców i samic (patrz punkt 5.3).
04.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Nie przeprowadzono badań dotyczących zdolności prowadzenia pojazdów i obsługiwania maszyn. Nie ma powodu, by sądzić, że sorafenib wpływa na zdolność prowadzenia pojazdów i obsługiwania maszyn.
04.8 Działania niepożądane
Najważniejszymi ciężkimi działaniami niepożądanymi były niedokrwienie i zawał mięśnia sercowego, perforacja przewodu pokarmowego, polekowe zapalenie wątroby, krwotok i nadciśnienie lub przełom nadciśnieniowy.
Najczęstszymi działaniami niepożądanymi były biegunka, osłabienie, łysienie, zakażenie, reakcja skórna dłoniowo-podeszwowa (odpowiadająca w MedDRA „erytrodyzestezji dłoniowo-podeszwowej”) oraz wysypka.
Działania niepożądane zgłaszane w różnych badaniach klinicznych lub po wprowadzeniu do obrotu są wymienione w tabeli 1, posortowane według MedDRA i częstości.
W każdej klasie częstotliwości działania niepożądane są przedstawione w porządku malejącym według stopnia nasilenia.
Tabela 1: Ogólne działania niepożądane zgłaszane u pacjentów w różnych badaniach klinicznych lub po wprowadzeniu do obrotu.
* Działania niepożądane mogą zagrażać życiu lub prowadzić do śmierci. Zdarzenia te są albo rzadkie, albo rzadsze niż rzadkie.
** Reakcja skórna ręka-stopa odpowiada zespołowi erytrodyzestezji dłoniowo-podeszwowej w MedDRA
Dowiedz się więcej o niektórych działaniach niepożądanych
Zastoinowa niewydolność serca
w sponsorowanym przez firmę badaniu klinicznym zastoinową niewydolność serca zgłoszono jako zdarzenie niepożądane u 1,9% pacjentów leczonych sorafenibem (n = 2276). W badaniu 11213 (RCC) zdarzenia niepożądane odpowiadające zastoinowej niewydolności serca zgłoszono u 1,7% pacjentów leczonych sorafenibem i u 0,7% pacjentów otrzymujących placebo. W badaniu 100554 (HCC) takie zdarzenia zgłoszono u 0,99% pacjentów leczonych sorafenibem i 1,1% pacjentów otrzymujących placebo.
Dodatkowe informacje dla populacji specjalnych
W badaniach klinicznych niektóre działania niepożądane, takie jak reakcja skórna ręka-stopa, biegunka, łysienie, utrata masy ciała, nadciśnienie, hipokalcemia i rogowiak kolczystokomórkowy/rak kolczystokomórkowy skóry, występowały ze znacznie większą częstością u pacjentów ze zróżnicowanym rakiem tarczycy w porównaniu z pacjentami. w badaniach nad rakiem nerki lub wątrobowokomórkowym.
Zmiany w badaniach laboratoryjnych u chorych na HCC (badanie 3) i RCC (badanie 1)
Bardzo często zgłaszano zwiększenie aktywności lipazy i amylazy Zwiększenie aktywności lipazy stopnia 3. lub 4. Wspólne kryteria toksycznościZdarzenia niepożądane (CTCAE) wystąpiły u 11% i 9% pacjentów w grupie sorafenibu odpowiednio w badaniu 1 (RCC) i badaniu 3 (HCC) w porównaniu z 7% i 9% pacjentów w grupie sorafenibu leczonych placebo. Zwiększenie aktywności amylazy stopnia 3. lub 4. wg CTCAE wystąpiło u 1% i 2% pacjentów w grupie sorafenibu odpowiednio w badaniu 1 i badaniu 3 w porównaniu do 3% pacjentów w obu grupach placebo. Kliniczne zapalenie trzustki zgłoszono u 2 z 451 pacjentów leczonych sorafenibem (stopień 4) wg CTCAE w badaniu 1, u 1 z 297 pacjentów leczonych sorafenibem (stopień 2) wg CTCAE w badaniu 3 i u 1 z 451 pacjentów (stopień 2) wg CTCAE, którym podawano placebo w badaniu 1.
Hipofosfatemia jest bardzo częstym objawem laboratoryjnym i była obserwowana odpowiednio u 45% i 35% pacjentów leczonych sorafenibem w badaniu 1 i badaniu 3 w porównaniu z odpowiednio 12% i 11% pacjentów otrzymujących placebo. Hipofosfatemia stopnia 3 wg CTCAE (1 - 2 mg/dl) wystąpiła w badaniu 1 u 13% pacjentów leczonych sorafenibem i u 3% pacjentów otrzymujących placebo, natomiast w badaniu 3 wystąpiła u 11% pacjentów leczonych sorafenibem i 2% pacjentów leczonych placebo Nie zgłoszono przypadków hipofosfatemii stopnia 4 wg CTCAE (etiologia hipofosfatemii związanej z sorafenibem jest nieznana).
Nieprawidłowości w wynikach badań laboratoryjnych stopnia 3. lub 4. wg CTCAE, w tym limfopenię i neutropenię, obserwowano u ≥ 5% pacjentów leczonych sorafenibem.
Hipokalcemię obserwowano u 12% i 26,5% pacjentów leczonych sorafenibem w porównaniu z 7,5% i 14,8% pacjentów w grupie placebo odpowiednio w badaniu 1 i 3. przypadki hipokalcemii były łagodne (stopień 1 i 2 wg CTCAE). Hipokalcemia stopnia 3 wg CTCAE (6,0 – 7,0 mg/dl) wystąpiła u 1,1% i 1,8% pacjentów leczonych sorafenibem oraz u 0,2% i 1,1% pacjentów w grupie placebo, a hipokalcemia stopnia 4 wg CTCAE (
W badaniach 1 i 3 zmniejszenie stężenia potasu zaobserwowano odpowiednio u 5,4% i 9,5% pacjentów leczonych sorafenibem w porównaniu z 0,7% i 5,9% pacjentów otrzymujących placebo. Większość przypadków hipokaliemii była łagodna (1 stopień wg CTCAE). W badaniach tych hipokaliemia stopnia 3. wg CTCAE wystąpiła u 1,1% i 0,4% pacjentów leczonych sorafenibem oraz u 0,2% i 0,7% pacjentów w grupie placebo. Przypadki hipokaliemii stopnia 4. wg CTCAE.
Zmiany w badaniach laboratoryjnych u pacjentów z DTC (badanie 5)
Hipokalcemię obserwowano u 35,7% pacjentów leczonych sorafenibem w porównaniu z 11,0% pacjentów w grupie placebo. Większość przypadków hipokalcemii miała łagodny przebieg. Hipokalcemia stopnia 3. wg CTCAE wystąpiła u 6,8% pacjentów leczonych sorafenibem i 1,9% pacjentów z grupy placebo, natomiast hipokalcemia stopnia 4. wg CTCAE wystąpiła u 3,4% pacjentów leczonych sorafenibem i u 1,0% pacjentów z grupy placebo.
Inne klinicznie istotne zmiany laboratoryjne obserwowane w badaniu 5 przedstawiono w Tabeli 2.
Tabela 2: Nieprawidłowości laboratoryjne związane z leczeniem zgłaszane u pacjentów z DTC (badanie 5) w fazie podwójnie ślepej próby
* Wspólne kryteria terminologiczne dotyczące zdarzeń niepożądanych (CTCAE), wersja 3.0
** Etiologia hipofosfatemii związanej z sorafenibem jest nieznana.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie podejrzewanych działań niepożądanych, które występują po dopuszczeniu produktu leczniczego do obrotu, jest ważne, ponieważ umożliwia ciągłe monitorowanie stosunku korzyści do ryzyka produktu leczniczego.Osoby należące do fachowego personelu medycznego proszone są o zgłaszanie wszelkich podejrzewanych działań niepożądanych za pośrednictwem strony internetowej Włoskiej Agencji Leków: www.agenziafarmaco.gov.it/it/responsabili.
04.9 Przedawkowanie
Nie ma specjalnych metod leczenia w przypadku przedawkowania sorafenibu. Najwyższa dawka sorafenibu badana klinicznie wynosi 800 mg dwa razy na dobę. Zdarzeniami niepożądanymi obserwowanymi po tej dawce były głównie biegunka i reakcje dermatologiczne. W przypadku podejrzenia przedawkowania sorafenib należy odstawić i, jeśli to konieczne, rozpocząć leczenie wspomagające.
05.0 WŁAŚCIWOŚCI FARMAKOLOGICZNE
05.1 Właściwości farmakodynamiczne
Grupa farmakoterapeutyczna: leki przeciwnowotworowe, inhibitory kinaz białkowych.
Kod ATC: L01XE05.
Sorafenib jest inhibitorem kinazy, który wykazał zarówno właściwości antyproliferacyjne, jak i antyangiogenne in vitro oraz in vivo.
Mechanizm działania i efekty farmakodynamiczne
Sorafenib jest inhibitorem kinazy hamującym proliferację komórek nowotworowych in vitro. Sorafenib hamuje wzrost szerokiego spektrum nowotworów ludzkich przeszczepionych myszom bez grasicy, co skutkuje również zmniejszeniem angiogenezy nowotworowej Sorafenib hamuje aktywność celów obecnych w komórce nowotworowej (CRAF, BRAF, V600E BRAF, c-KIT i FLT- 3) oraz w naczyniach krwionośnych guza (CRAF, VEGFR-2, VEGFR-3 i PDGFR-Ÿ). Kinazy RAF to kinazy serynowo-treoninowe, podczas gdy c-KIT, FLT-3, VEGFR-2, VEGFR-3 i PDGFR-ß to receptorowe kinazy tyrozynowe.
Skuteczność kliniczna
Bezpieczeństwo i skuteczność kliniczną sorafenibu badano u pacjentów z rakiem wątrobowokomórkowym (rak wątrobowokomórkowy, HCC), u pacjentów z zaawansowanym rakiem nerkowokomórkowym (rak nerkowokomórkowy, RCC) oraz u pacjentów ze zróżnicowanym rakiem tarczycy (zróżnicowany rak tarczycy, DTC).
rak wątroby
Badanie 3 (badanie 100554) było wieloośrodkowym, randomizowanym, prowadzonym metodą podwójnie ślepej próby, kontrolowanym placebo, międzynarodowym badaniem klinicznym III fazy z udziałem 602 pacjentów z rakiem wątrobowokomórkowym. Wyjściowe dane demograficzne i charakterystyka choroby były porównywalne między grupami sorafenibu i placebo w odniesieniu do klasyfikacji Estern Cooperative Oncology Group (ECOG) (stopień 0: 54% versus 54%; stopień 1: 38% versus 39%; stopień 2: 8% versus 7%), do klasyfikacji TNM (etap I:
Badanie zostało zamknięte po tym, jak zaplanowana śródokresowa analiza całkowitego przeżycia (OS) przekroczyła wstępnie zdefiniowany limit skuteczności. Ta analiza OS wykazała statystycznie istotny wzrost OS dla pacjentów leczonych sorafenibem w porównaniu z pacjentami otrzymującymi placebo (HR: 0,69, p = 0,00058, patrz tabela 3).
W tym badaniu dane dotyczące pacjentów z zaburzeniami czynności wątroby w skali Child Pugh B są ograniczone i uwzględniono tylko jednego pacjenta w skali Child Pugh C.
Tabela 3: Wyniki skuteczności z badania 3 (badanie 100554) w raku wątroby
CI = Przedział ufności, HR = Współczynnik ryzyka (sorafenib w porównaniu z placebo)
* statystycznie istotna, ponieważ wartość p była mniejsza niż domyślna granica odcięcia O „Brien Fleming, ustalona na 0,0077
** niezależny przegląd radiologiczny
W drugim, międzynarodowym, wieloośrodkowym, randomizowanym, podwójnie zaślepionym, kontrolowanym placebo badaniu fazy III (badanie 4, 11849) oceniano korzyści kliniczne sorafenibu u 226 pacjentów z zaawansowanym rakiem wątroby. Badanie to, przeprowadzone w Chinach, Korei i na Tajwanie, potwierdziło wyniki Badania 3 w odniesieniu do korzystnego profilu korzyści do ryzyka sorafenibu (HR (OS): 0,68, p = 0,01414).
We wstępnie zdefiniowanych czynnikach stratyfikacji (klasyfikacja ECOG, obecność lub brak makroskopowego naciekania naczyń i (lub) pozawątrobowe rozprzestrzenienie się guza) z badań 3 i 4, HR konsekwentnie opowiadał się za sorafenibem w porównaniu z placebo. Analizy eksploracyjne podgrup sugerowały mniej wyraźny efekt leczenia u pacjentów z przerzutami odległymi już na początku badania.
Rak nerkowokomórkowy
Tolerancję i skuteczność sorafenibu w leczeniu zaawansowanego raka nerkowokomórkowego (RCC) badano w dwóch badaniach klinicznych:
Badanie 1 (badanie 11213) było wieloośrodkowym, randomizowanym, podwójnie zaślepionym, kontrolowanym placebo badaniem klinicznym III fazy, przeprowadzonym z udziałem 903 pacjentów. Do badania włączono wyłącznie pacjentów z jasnokomórkowymi guzami nerki iz niskim i średnim czynnikiem ryzyka wg MSKCC. ten punkt końcowypierwotne były przeżycie całkowite (OS, ogólnie przetrwanie) i przeżycia bez progresji choroby (PFS, Postęp Darmowy Przetrwanie).
Około połowa pacjentów miała stan ogólny równy 0 w skali ECOG, a połowa pacjentów należała do grupy prognostycznej z niskim wynikiem według klasyfikacji MSKCC.
PFS oceniano zgodnie z kryteriami RECIST z zaślepioną niezależną oceną radiologiczną. Analizę PFS przeprowadzono dla 342 zdarzeń u 769 pacjentów Mediana wartości PFS wyniosła 167 dni u pacjentów leczonych sorafenibem w porównaniu z 84 dniami u pacjentów otrzymujących placebo (HR = 0,44; 95% CI: 0,35 - 0,55; p
„Analiza okres przejściowy (druga analiza okres przejściowy) dla całkowitego przeżycia (ogólnie przetrwanie) przeprowadzono przy 367 zgonach u 903 pacjentów. Nominalna wartość alfa dla tej analizy wyniosła 0,0094. Mediana przeżycia wyniosła 19,3 miesiąca u pacjentów leczonych sorafenibem w porównaniu z 15,9 miesiąca u pacjentów randomizowanych do grupy placebo (HR = 0,77; 95% CI: 0,63-0,95; p = 0,015). W momencie analizy około 200 pacjentów przeszło z grupy placebo do grupy sorafenibu.
Badanie 2 było badaniem fazy II, w którym randomizowano przerwanie leczenia u pacjentów z rakiem z przerzutami, w tym z rakiem nerkowokomórkowym. Pacjenci ze stabilizacją choroby i leczeni sorafenibem byli randomizowani do grupy otrzymującej placebo lub do kontynuacji leczenia sorafenibem. PFS u pacjentów z rakiem nerkowokomórkowym był znacznie większy [163]. dni) dla pacjentów leczonych sorafenibem niż obserwowane u pacjentów otrzymujących placebo (41 dni) (p = 0,0001, HR = 0,29).
Zróżnicowany rak tarczycy (DTC)
Badanie 5 (badanie 14295) było międzynarodowym, wieloośrodkowym, randomizowanym, podwójnie zaślepionym, kontrolowanym placebo badaniem III fazy, przeprowadzonym z udziałem 417 pacjentów z miejscowo zaawansowanym lub przerzutowym DTC opornym na jod promieniotwórczy. Przeżycie wolne od progresji choroby (PFS), określone na podstawie zaślepionej niezależnej oceny radiologicznej w oparciu o kryteria RECIST, było pierwszorzędowym punktem końcowym badania.Drugorzędowe punkty końcowe obejmowały przeżycie całkowite (OS), odsetek odpowiedzi guza i czas trwania odpowiedzi Po progresji pacjenci mogli otrzymać sorafenib w ramach otwartej próby.
Pacjenci byli włączani do badania, jeśli mieli progresję w ciągu 14 miesięcy przed włączeniem i jeśli mieli DTC oporny na radiojod (radioaktywny jod, RAI). DTC oporny na RAI zdefiniowano jako obecność zmiany niewzmagającej jodu w scyntygrafii RAI lub łączne podanie RAI ≥ 22,2 GBq lub progresję po leczeniu RAI w ciągu ostatnich 16 miesięcy. maksymalna odległość 16 miesięcy od siebie.
Wyjściowa demografia i charakterystyka pacjentów były dobrze zrównoważone w dwóch grupach terapeutycznych. Przerzuty były obecne w płucach w 86%, w węzłach chłonnych w 51% iw kościach u 27% chorych. Mediana skumulowanej aktywności radiojodu podanej przed włączeniem wynosiła około 14,8 GBq. Większość pacjentów miała raka brodawkowatego (56,8%), następnie raka pęcherzykowego (25,4%) i raka słabo zróżnicowanego (9,6%).
Mediana czasu do PFS wyniosła 10,8 miesiąca w grupie sorafenibu w porównaniu z 5,8 miesiąca w grupie placebo. (HR = 0,587; 95% przedział ufności (CI): 0,454; 0,758; p jednostronne
Wpływ sorafenibu na PFS był stały niezależnie od regionu geograficznego, wieku powyżej lub poniżej 60 lat, płci, histologii oraz obecności lub braku przerzutów do kości.
W analizie przeżycia całkowitego przeprowadzonej 9 miesięcy po dacie odcięcia dla końcowej analizy PFS, nie było statystycznie istotnej różnicy w całkowitym przeżyciu między grupami leczenia (HR 0,884; CI 95%: 0,633; 1,236, jednostronne p- wartość 0,236). Mediana OS nie została osiągnięta w ramieniu sorafenibu, podczas gdy w ramieniu placebo wyniosła 36,5 miesiąca Sto pięćdziesiąt siedem pacjentów (75%) randomizowanych do grupy placebo i 61 pacjentów (30%) randomizowanych do grupy otrzymującej sorafenib otrzymywało sorafenib metodą otwartej próby.
Mediana czasu trwania leczenia w fazie podwójnie ślepej próby wyniosła 46 tygodni (zakres 0,3-135) u pacjentów otrzymujących sorafenib i 28 tygodni (zakres 1,7-132) u pacjentów otrzymujących placebo.
Nie zaobserwowano pełnej odpowiedzi (pełna odpowiedź, CR) według kryteriów RECIST. Całkowity odsetek odpowiedzi (CR + odpowiedź częściowa, częściowa odpowiedź (PR)), określony w niezależnej ocenie radiologicznej, był wyższy w grupie sorafenibu (24 pacjentów, 12,2%) w porównaniu z grupą placebo (1 pacjent, 0,5%), p jednostronny
Analiza post-post podgrup w oparciu o maksymalną wielkość guza wykazała wpływ leczenia na PFS na korzyść sorafenibu w porównaniu z placebo u pacjentów z maksymalną wielkością zmiany guza 1,5 cm lub większą (HR 0,54 (95% CI: 0,41-0,71)). , podczas gdy liczbowo mniejszy efekt odnotowano u pacjentów z maksymalną wielkością zmiany guza mniejszą niż 1,5 cm (HR 0,87 (95% CI: 0,40 -1,89)).
Analiza post-hoc oparta na objawach związanych z rakiem tarczycy występujących w warunkach wyjściowych wykazała wpływ leczenia na PFS na korzyść sorafenibu w porównaniu z placebo zarówno u pacjentów z objawami, jak i bez objawów.HR dla progresji wolnego czasu przeżycia wyniósł 0,39 (95% CI: 0,21 - 0,72) dla pacjentów z objawami na początku i 0,60 (95% CI: 0,45 - 0,81) dla pacjentów bez objawów w warunkach podstawowych.
Wydłużenie odstępu QT
W badaniu farmakologii klinicznej QT/QTc mierzono u 31 pacjentów na początku leczenia (przed leczeniem) i po leczeniu. Po 28-dniowym cyklu leczenia, w momencie maksymalnego stężenia sorafenibu, QTcB wydłużył się o 4 ± 19 ms, a QTcF o 9 ± 18 ms, w porównaniu z wartością wyjściową dla grupy placebo. Żaden pacjent nie wykazywał wartości QTcB lub QTcF > 500 ms podczas monitorowania EKG po leczeniu (patrz punkt 4.4).
Populacja pediatryczna
Europejska Agencja Leków uchyliła obowiązek przedłożenia wyników badań we wszystkich podgrupach populacji pediatrycznej z rakiem nerki i miedniczki nerkowej (z wyjątkiem nerczaka niedojrzałego, nefroblastomatozy, mięsaka jasnokomórkowego, nerka mezoblastycznego, raka rdzeniastego nerki i guza rabdoidalnego nerki) oraz rak wątroby i wewnątrzwątrobowych dróg żółciowych (z wyjątkiem wątrobiaka zarodkowego) oraz zróżnicowany rak tarczycy (informacje dotyczące stosowania u dzieci, patrz punkt 4.2).
05.2 „Właściwości farmakokinetyczne
Wchłanianie i dystrybucja
Po podaniu tabletek sorafenibu średnia względna biodostępność wynosi 38-49% w porównaniu z roztworem doustnym. Całkowita biodostępność jest nieznana. Po podaniu doustnym sorafenib osiąga maksymalne stężenie w osoczu po około 3 godzinach. Po podaniu z posiłkiem o dużej zawartości tłuszczu wchłanianie sorafenibu jest zmniejszone o około 30% w porównaniu do podawania na czczo.
Średnie Cmax i AUC wzrastają mniej niż proporcjonalnie przy dawkach powyżej 400 mg dwa razy na dobę. Wiązanie sorafenibu z białkami osocza in vitro wynosi 99,5%.
Wielokrotne podawanie sorafenibu przez 7 dni powodowało od 2,5 do 7-krotną kumulację w porównaniu z podaniem pojedynczym. Stan stacjonarny sorafenibu osiągany jest w ciągu 7 dni, ze stosunkiem średniego stężenia maksymalnego do minimalnego w osoczu poniżej 2.
Stężenia równowagowe sorafenibu podawanego w dawce 400 mg dwa razy na dobę oznaczono u pacjentów z DTC, RCC i HCC, przy czym najwyższe średnie stężenie obserwowano u pacjentów z DTC (około dwukrotnie większe niż obserwowane u pacjentów z RCC i HCC), ale zmienność była duża dla wszystkich typów nowotworów Przyczyna tego wyższego stężenia u pacjentów z DTC jest nieznana.
Biotransformacja i eliminacja
Okres półtrwania sorafenibu w fazie eliminacji wynosi około 25 do 48 godzin. Sorafenib jest metabolizowany głównie w wątrobie poprzez metabolizm oksydacyjny za pośrednictwem CYP3A4 i sprzęganie glukurono za pośrednictwem UGT1A9. Sprzężony sorafenib może być uwalniany w przewodzie pokarmowym przez aktywność glukuronidazy niektórych bakterii, umożliwiając w ten sposób reabsorpcję nieskoniugowanej substancji czynnej.Zaobserwowano, że połączenie z neomycyną zakłóca ten proces, zmniejszając średnią biodostępność sorafenibu o 54%.
Sorafenib stanowi około 70-85% analitów krążących w osoczu w stanie stacjonarnym. Zidentyfikowano osiem metabolitów sorafenibu, z których pięć znaleziono w osoczu. Główny metabolit sorafenibu krążącego w osoczu, N-tlenek pirydyny, wykazuje silne działaniein vitro podobny do sorafenibu. Metabolit ten stanowi około 9-16% analitów krążących w stanie stacjonarnym.
Po podaniu doustnym dawki 100 mg roztworu sorafenibu 96% dawki odzyskano w ciągu 14 dni: 77% w kale i 19% w moczu w postaci metabolitów glukuronianowych. Niezmieniony sorafenib, stanowiący 51% dawki, był wydalany z kałem, ale nie z moczem, co wskazuje, że wydalanie niezmetabolizowanej substancji czynnej z żółcią może przyczyniać się do eliminacji sorafenibu.
Farmakokinetyka w poszczególnych kategoriach pacjentów
Analiza danych demograficznych wykazała, że nie ma korelacji pomiędzy farmakokinetyką a wiekiem (do 65 lat), płcią czy masą ciała.
Populacja pediatryczna
Nie przeprowadzono badań w celu weryfikacji farmakokinetyki sorafenibu u dzieci.
Wyścigi
Nie ma klinicznie istotnych różnic w farmakokinetyce między osobami rasy kaukaskiej i azjatyckiej.
Zaburzenia czynności nerek
W czterech badaniach klinicznych fazy I ekspozycja na sorafenib w stanie stacjonarnym u pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności nerek była podobna do obserwowanej u pacjentów z prawidłową czynnością nerek.W badaniu farmakologii klinicznej (pojedyncza dawka 400 mg sorafenibu) nie zaobserwowano związku między ekspozycją na sorafenib a czynnością nerek u osób z prawidłową czynnością nerek lub łagodnymi, umiarkowanymi lub ciężkimi zaburzeniami czynności nerek. Brak danych dotyczących pacjentów wymagających dializy.
Zaburzenia czynności wątroby
U pacjentów z rakiem wątrobowokomórkowym (HCC) oraz z zaburzeniami czynności wątroby ocenianymi jako A lub B w skali Child-Pugh (łagodne do umiarkowanych), wartości ekspozycji były porównywalne i mieściły się w zakresie obserwowanym u pacjentów bez zaburzeń czynności wątroby. Farmakokinetyka sorafenibu u pacjentów A i B w skali Child-Pugh bez HCC była podobna do obserwowanej u zdrowych ochotników. Brak danych dotyczących pacjentów z ciężkimi zaburzeniami czynności wątroby (C w skali Childa-Pugha). Sorafenib jest eliminowany głównie przez wątrobę i w tej populacji pacjentów ekspozycja może być zwiększona.
05.3 Przedkliniczne dane o bezpieczeństwie
Przedkliniczny profil bezpieczeństwa sorafenibu oceniano u myszy, szczurów, psów i królików.
Badania toksyczności po podaniu wielokrotnym ujawniły zmiany w różnych narządach (degeneracja i regeneracja) przy ekspozycji mniejszej niż dawki stosowane w badaniach klinicznych (na podstawie porównania AUC).
Po wielokrotnym dawkowaniu młodym i rosnącym psom zaobserwowano wpływ na kości i zęby przy ekspozycji mniejszej niż dawki stosowane w badaniach klinicznych. Efekty te polegały na nierównomiernym pogrubieniu chrząstki wzrostowej kości udowej, hipoplazji rdzenia kręgowego w sąsiedztwie zmienionych chrząstek wzrostowych oraz zmianach w składzie zębiny. Podobnych efektów nie wywołano u dorosłego psa.
Przeprowadzono standardowy program badań genotoksyczności i uzyskano pozytywne wyniki, ponieważ w jednym teście odnotowano wzrost aberracji strukturalnych chromosomów. in vitro w komórkach ssaków (jajniki chomika chińskiego) do pomiaru klastogenności w obecności aktywacji metabolicznej. Sorafenib nie był genotoksyczny w teście Amesa ani teście mikrojądrowym in vivo w myszy. Półprodukt procesu produkcyjnego, który jest również obecny w końcowej substancji czynnej (in vitro na komórkach bakteryjnych (test Amesa). Ponadto partia sorafenibu badana w standardowej baterii genotoksycznej zawierała 0,34% PAPE.
Nie przeprowadzono badań rakotwórczości sorafenibu.
Nie przeprowadzono specjalnych badań sorafenibu na zwierzętach w celu oceny wpływu na płodność.Należy jednak spodziewać się niekorzystnego wpływu na płodność samców i samic, ponieważ badania na zwierzętach z wielokrotnym podawaniem dawek wykazały zmiany w męskich i żeńskich narządach rozrodczych przy ekspozycji mniejszej niż dawki stosowane w badaniach klinicznych (na podstawie AUC). oznaki zwyrodnienia i opóźnionego rozwoju jąder, najądrza, prostaty i pęcherzyków nasiennych u szczurów samice szczurów wykazywały centralną martwicę ciał żółtych i blokadę rozwoju pęcherzyków jajnikowych psy wykazały zwyrodnienie kanalików w jajnikach jądra i oligospermię
Wykazano, że sorafenib ma działanie embriotoksyczne i teratogenne, gdy jest podawany szczurom i królikom przy ekspozycji mniejszej niż dawki stosowane w badaniach klinicznych. Obserwowane efekty obejmowały zmniejszenie masy ciała matki i płodu, wzrost liczby resorpcji płodu oraz zwiększoną liczbę wad zewnętrznych i trzewnych.
Badania oceny ryzyka środowiskowego wykazały, że tosylan sorafenibu jest potencjalnie trwały, zdolny do bioakumulacji i toksyczny dla środowiska.Informacje na temat oceny ryzyka środowiskowego są dostępne w Europejskim Publicznym Sprawozdaniu Oceniającym (EPAR) dla tego produktu leczniczego (patrz punkt 6.6).
06.0 INFORMACJE FARMACEUTYCZNE
06.1 Zaróbki
Rdzeń tabletu:
Kroskarmeloza sodowa
Celuloza mikrokrystaliczna
Hypromeloza
Laurylosiarczan sodu
Stearynian magnezu
Powłoka tabletu:
Hypromeloza
Makrogol
Dwutlenek tytanu (E 171)
Czerwony tlenek żelaza (E 172)
06.2 Niezgodność
Nieistotne.
06.3 Okres ważności
3 lata.
06.4 Specjalne środki ostrożności przy przechowywaniu
Nie przechowywać w temperaturze powyżej 25°C.
06.5 Rodzaj opakowania bezpośredniego i zawartość opakowania
Pudełko tekturowe zawierające 112 tabletek powlekanych (4 x 28) w przezroczystym blistrze (PP/Aluminium).
06.6 Instrukcje użytkowania i obsługi
Ten produkt leczniczy może stanowić potencjalne zagrożenie dla środowiska.Niezużyty produkt leczniczy i odpady pochodzące z tego produktu leczniczego należy usunąć zgodnie z lokalnymi przepisami.
07.0 PODMIOT POZWOLENIA NA DOPUSZCZENIE DO OBROTU
Bayer Pharma AG
13342 Berlin
Niemcy
08.0 NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
UE / 1/06/342/001
037154010
09.0 DATA PIERWSZEGO ZEZWOLENIA LUB PRZEDŁUŻENIA ZEZWOLENIA
Data pierwszego zezwolenia: 19 lipca 2006 r.
Data ostatniego przedłużenia: 21 lipca 2011 r.
10.0 DATA ZMIAN TEKSTU
05/2014