Ogólność
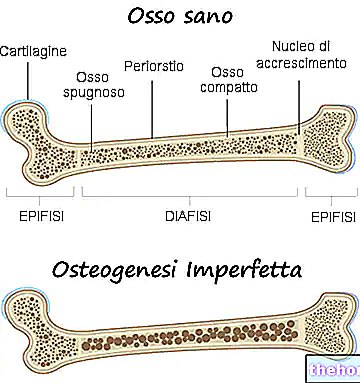
Osteogenesis imperfecta jest wrodzoną chorobą genetyczną niezwiązaną z płcią, odpowiedzialną za pewną kruchość kości i wyraźną skłonność do złamań.
Objawy imperfecta osteogenezy są liczne, na ogół składają się na nie: osłabienie kości, duża skłonność do złamań kości, obecność niebieskich, szarych lub fioletowych twardówek ocznych, obecność deformacji kości lub innych zmian kostnych, trójkątna twarz, kruchość zębów itp. .
Zasadniczo do prawidłowego rozpoznania wady wrodzonej osteogenezy niezbędne są: badanie fizykalne, wywiad chorobowy, badania obrazowe, ocena kolagenu typu I oraz badanie genetyczne.
Niestety obecnie jedyne metody leczenia dostępne dla pacjentów z wrodzoną łamliwością kości są objawowe. Omawiana choroba jest w rzeczywistości nieuleczalna.
Czym jest wrodzona wada osteogenezy?
Osteogenesis imperfecta jest chorobą genetyczną, która sprawia, że kości chorego są słabsze i bardziej podatne na złamania.
W rzeczywistości, terminem osteogeneza imperfecta, lekarze odnoszą się do niejednorodnej grupy chorób genetycznych, charakteryzujących się pewnym stopniem kruchości kości. Istnieje zatem kilka postaci (lub typów) wady wrodzonej osteogenezy, niektóre znacznie cięższe niż inne.
TO JEST WRODZONA CHOROBA
U osób nią dotkniętych osteogenesis imperfecta jest chorobą występującą od urodzenia, dlatego można ją zdefiniować jako chorobę wrodzoną.
CZY TO ZWIĄZANE Z SEKSEM?
Osteogenesis imperfecta nie jest chorobą genetyczną związaną z płcią, taką jak hemofilia czy zespół Klinefeltera.
EPIDEMILOGIA
Według niektórych badań statystycznych zapadalność na wrodzoną łamliwość kości byłaby równa jednemu przypadkowi na 15 000-20 000 urodzeń. Oznacza to, że co 15 000-20 000 noworodków ma jeden dotknięty wrodzoną wadą osteogenezy.
Inne badania statystyczne wykazały również, że osteogeneza wrodzona w równym stopniu dotyka mężczyzn i kobiety oraz że nie ma preferencji dla określonej populacji lub grupy etnicznej.
Długość życia jest niezwykle zmiennym parametrem, który zależy od formy wady wrodzonej osteogenezy.
Powoduje
Osteogenesis imperfecta prawie zawsze wynika z jakościowej i ilościowej zmiany produkcji kolagenu typu I.
Kolagen typu I jest niezbędny do wzmocnienia kości i utrzymania zdrowych tkanek łącznych stanowiących chrząstki, ścięgna, skórę, twardówkę oka itp.
Dlatego zmiana w produkcji kolagenu typu I wpływa na wytrzymałość kości i dobry stan zdrowia tkanek łącznych obecnych w ludzkim ciele.
CO ZMIENIA PRODUKCJĘ KOLAGENU?
Choroba genetyczna to stan, który powstaje w wyniku mutacji jednego lub więcej genów tworzących DNA komórki.
W przypadku wrodzonej osteogenezy przyczyny tej ostatniej prawie zawsze można znaleźć w mutacji jednego lub obu genów COL1A1 (zlokalizowanych na chromosomie 17) i COL1A2 (zlokalizowanych na chromosomie 7).
W normalnych warunkach COL1A1 i COL1A2 regulują normalną produkcję kolagenu typu I; w obecności mutacji w swoim podopiecznym zawodzą w ich funkcji regulacyjnej.
Ważne: jakie inne geny, jeśli są zmutowane, powodują wrodzoną osteogenezę?
Oprócz mutacji COL1A1 i COL1A2 potencjalną przyczyną wady osteogenezy są mutacje w genach IFITM5, SERPINF1, CRTAP i LEPRE1.
Wspomniane geny pokrywają inne funkcje niż COL1A1 i COL1A2 – dlatego nie kontrolują produkcji kolagenu typu I – ale nadal mają „wpływ na wytrzymałość i odporność kości szkieletu człowieka”.
CO TO JEST CHOROBA GENETYCZNA?
Osteogenesis imperfecta jest autosomalną chorobą genetyczną.
Termin autosomalny, związany z chorobą genetyczną, wskazuje, że stan, o którym mowa, jest spowodowany mutacjami genetycznymi opartymi na chromosomach autosomalnych i niepłciowych.
Czytelnikom przypomina się, że człowiek posiada zestaw chromosomów składający się z 23 par chromosomów całkowitych, z czego 22 pary są typu autosomalnego, a tylko jedna jest typu płciowego.Para chromosomów typu płciowego wpływa na płeć indywidualny .
Osteogenesis imperfecta w następstwie mutacji w COL1A1, COL1A2 i IFITM5 ma wszystkie cechy choroby autosomalnej dominującej, a w przypadku mutacji w genach SERPINF1, CRTAP i LEPRE1 ma cechy choroby autosomalnej recesywnej.
TYPY
Obecnie lekarze uważają, że istnieje 8 rodzajów (lub form) wady wrodzonej osteogenezy. Aby rozróżnić poszczególne typy, zdecydowali się na użycie numeracji rzymskiej, a dokładnie pierwszych ośmiu cyfr rzymskich.
Poniższa tabela przedstawia 8 form wady osteogenezy, mutacje, które je wywołują oraz inne cechy.
Facet
Zmutowany gen
Rodzaj choroby genetycznej
TEN
COL1A1
Autosomalna dominująca
II
COL1A1 i COL1A2
Autosomalna dominująca
III
COL1A1 i COL1A2
Autosomalna dominująca
IV
COL1A1 i COL1A2
Autosomalna dominująca
V.
IFITM5
Autosomalna dominująca
TY
SERPINF1
Autosomalny recesywny
VII
CRTAP
Autosomalny recesywny
VIII
ZAJĄC 1
Autosomalny recesywny
* Uwaga: oczywiście mutacje w COL1A1 i COL1A2, które powodują pierwsze cztery formy wady osteogenezy, są zmianami genetycznymi o nieco innych cechach. W przeciwnym razie rozróżnianie jednego od drugiego nie miałoby sensu.
Objawy, oznaki i powikłania
Wszystkie rodzaje wady osteogenezy są odpowiedzialne za osłabienie kości, tak że osoba dotknięta chorobą ma szczególną predyspozycje do złamań. Stopień osłabienia kości zmienia się w zależności od kształtu; dla niektórych z nich osłabienie to jest większe niż dla innych.
Powiedziawszy to, należy podkreślić, że każda forma wady osteogenezy ma swój własny obraz objawowy, który dla niektórych może przypominać obraz symptomatologiczny innych form.
MOŻLIWE OBJAWY I OZNAKI
Możliwe objawy i oznaki wady osteogenezy obejmują:
- Obecność wad rozwojowych kości;
- Obecność krótkiego i małego ciała (przeznaczonego jako tułów);
- Problemy ze stawami (np. luźne stawy);
- Słabe mięśnie;
- Niebieska, fioletowa lub szara twardówka oka;
- Trójkątna twarz;
- Skrzynia z beczki;
- Anomalie morfologiczne kręgosłupa;
- kruchość zębów;
- Spadek lub całkowita utrata słuchu;
- Problemy z oddychaniem
- Problemy związane z brakiem lub brakiem kolagenu typu 1.
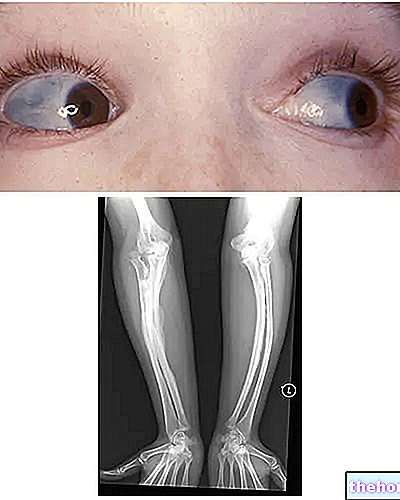
Osteogenesis Imperfecta: zwróć uwagę na niebieskie zabarwienie twardówki i deformacje kości, które charakteryzują tę chorobę. Z wikipedia.org
JAKIE SĄ NAJCIĘŻSZE FORMY NIEDOSKONAŁEJ OSTEOGENEZY?
Lekarze klasyfikują nasilenie symptomatologiczne różnych typów wady wrodzonej osteogenezy w skali 3 stopni, którymi są: stopień łagodny, stopień umiarkowany i stopień ciężki.
Tylko jedna forma należy do kategorii „łagodny stopień”: „typ I osteogenesis imperfecta”; 4 formy osteogenesis imperfecta należą do kategorii „umiarkowanego stopnia”: IV, V i VI; wreszcie do kategorii „ciężkiego stopnia” należą 3 formy: II, III, VII i VIII.
TYP I: CECHY
Najbardziej powszechna i najmniej ciężka forma ze wszystkich, osteogeneza niedoskonałości typu I, ma następujące cechy:
- Powoduje złamania, zwłaszcza przed okresem dojrzewania;
- Nie ma „prawie żadnego wpływu na wzrost, więc pacjenci mają zwykle normalny” wzrost;
- Powoduje problemy ze stawami i osłabienie mięśni
- Odpowiada za niebieską, fioletową lub szarą twardówkę;
- Jest przyczyną trójkątnych anomalii twarzy i kręgosłupa;
- Prawie nigdy nie powoduje deformacji kości. Jeśli to ich prowokuje, są minimalne;
- Może powodować kruchość zębów i / lub utratę słuchu (ta ostatnia zwykle występuje w wieku dorosłym);
- Jest to związane z obecnością kolagenu typu I, który jest normalnej jakości, ale nienormalnej ilości (jest uboższy niż normalnie).
TYP II: CECHY
Wrodzona osteogeneza typu II charakteryzuje się:
- Przyczyna zgonu przy urodzeniu lub wkrótce potem. Problemy z oddychaniem prawie zawsze powodują śmierć;
- Obecność znacznej kruchości kości i poważnych deformacji kości;
- Niski wzrost i słabo rozwinięte płuca
- Twardówka koloru niebieskiego, fioletowego lub szarego;
- Obecność anomalii ilościowych i jakościowych kolagenu typu I.
TYP III: CECHY
Wrodzona niedoskonałość osteogenezy typu III ma następujące cechy:
- Choć bardzo poważna, nie powoduje często śmierci w okresie noworodkowym;
- Jest to związane z „wysoką kruchością kości;
- Odpowiada za niski wzrost, problemy ze stawami, osłabienie mięśni (zwłaszcza nóg i ramion), beczkowatą klatkę piersiową, trójkątną twarz i nieprawidłową krzywiznę kręgosłupa;
- Jest to spowodowane niebieską, fioletową lub szarą twardówką;
- Może powodować problemy z oddychaniem, kruchość zębów i utratę słuchu;
- Często odpowiada za deformacje kości;
- Wiąże się z nieprawidłowościami jakościowymi i ilościowymi kolagenu typu I.
TYP IV: CECHY
Osteogeneza typu IV charakteryzuje się:
- Stopień kruchości kości między formami II i III a formą I;
- Niższy niż przeciętny wzrost;
- Twardówka koloru niebieskiego, fioletowego lub szarego;
- Deformacje kości łagodne / umiarkowane, niewielkie nieprawidłowości kręgosłupa i baryłkowatej klatki piersiowej;
- Trójkątna twarz;
- Możliwa obecność kruchości zębów i utraty słuchu;
- Obecność nieprawidłowości kolagenu typu I.
TYP V: CECHY
Osteogenesis imperfecta typu V przypomina pod pewnymi względami osteogenezę imperfecta typu IV. Ma jednak pewne osobliwości, którymi są:
- Normalnie zabarwiona twardówka;
- Brak kruchości zębów;
- Powstawanie nieprawidłowych modzeli kostnych podczas procesu gojenia złamanych kości;
- Zwapnienie błony międzykostnej znajdującej się między kością promieniową a kością łokciową. To upośledza ruchomość przedramienia.
TYP VI: CECHY
Również osteogenesis imperfecta typu VI jest podobna do formy IV. Odróżnić ją od tego ostatniego są pewne osobliwości, w tym wysoki poziom fosfatazy alkalicznej we krwi i obecność na niektórych kościach blaszek (kości) podobnych do kolców ryb.
TYP VII: CECHY
Symptomatycznie wrodzona wrodzona osteogeneza typu VII może w pewnych okolicznościach przypominać typ IV, a w innych typ II.
Osobliwości tej poważnej formy patologicznej obejmują:
- Niski wzrost;
- Obecność bardzo krótkiej kości ramiennej (kości ramienia) i kości udowej (kości udowej);
- Częsta obecność deformacji biodra, znanej jako coxa vara.
TYP VIII: CHARAKTERYSTYKA
Wrodzona wrodzona osteogeneza typu VIII bardzo przypomina formy II i III.
Wśród jego szczególnych cech wyróżniają się: silny deficyt wzrostu, silna hipomineralizacja szkieletu i brak (lub niewielka obecność) enzymu 3-hydroksylazy prolilowej.
Diagnoza
Ogólnie proces diagnostyczny, któremu poddawani są pacjenci z podejrzeniem wady wrodzonej osteogenezy, rozpoczyna się od dokładnego badania fizykalnego i dokładnego wywiadu lekarskiego; następnie kontynuuje się „analizą historii rodzinnej pacjenta i serią badań diagnostycznych obrazowych (zdjęcia rentgenowskie, tomografia komputerowa itp.); wreszcie kończy się ilościową i jakościową oceną kolagenu typu I oraz test genetyczny.
Obecnie istnieje możliwość diagnozowania wady osteogenezy już w fazie prenatalnej poprzez poddanie ciężarnej USG.
ZNACZENIE BADANIA CELOWEGO I HISTORIA
Lekarz specjalista w zakresie osteogenezy niedoskonałości bardzo często jest w stanie zdiagnozować wspomnianą chorobę nawet tylko na podstawie badania fizykalnego i wywiadu. Oznacza to, że te testy diagnostyczne nie mają bez znaczenia.
OCENA PRODUKCJI KOLAGENU TYPU I
Z reguły jakościowa i ilościowa ocena kolagenu typu I jest badaniem bardzo wiarygodnym, ponieważ, jak stwierdzono, większość przypadków wrodzonej osteogenezy charakteryzuje się mutacjami w genach kontrolujących produkcję kolagenu typu 1.
Aby ocenić ilość i jakość kolagenu typu I obecnego na poziomie komórkowym u osobnika, lekarze mogą polegać na biopsji skóry lub określonym badaniu krwi.
Oba te testy oceniające są dość złożone i pacjent (lub jego rodzice) może czekać kilka tygodni na poznanie wyników.
TEST GENETYCZNY
Dzięki testowi genetycznemu, który bada całe DNA badanej osoby, lekarze są w stanie definitywnie określić charakterystykę obecnej mutacji genetycznej.
Generalnie, wykonanie testu genetycznego na całym komórkowym DNA jest przewidziane, gdy ocena właściwości kolagenu typu I nie dała pożądanych wyników lub gdy nie jest to mutacja w COL1A1 lub COL1A2 powodująca wrodzoną osteogenezę.
DIAGNOZA PRENATALNA
Ultrasonografia prenatalna jest bardzo przydatna w identyfikacji niedoskonałości osteogenezy typu II i typu III.
Terapia
Obecnie nie ma konkretnego lekarstwa na wrodzoną wadę osteogenezy, innymi słowy osoby z wrodzoną wadą osteogenezy są skazani na dożycie z wyżej wymienionym stanem aż do śmierci, co często wynika z konsekwencji samej choroby.
Brak specyficznej terapii nie wyklucza istnienia innych form leczenia. W rzeczywistości, wśród możliwości terapeutycznych pacjenta z wrodzoną łamliwością kości, znajdują się różne terapie objawowe; przez terapie objawowe rozumiemy terapie zdolne do łagodzenia objawów, spowalniania przebiegu choroby i zapobiegania (lub przynajmniej opóźniania) najpoważniejszych konsekwencji.
MOŻLIWE ZABIEGI OBJAWOWE
Na liście możliwych sposobów leczenia objawowego wady wrodzonej osteogenezy wyróżniają się:
- Chirurgiczne wprowadzenie w najdłuższe kości (notabene: najbardziej podatne na złamania) gwoździ, które zapewniają większą odporność na złamania i deformacje. Ta operacja nazywa się rodding śródszpikowy;
- Zachowawcze lub chirurgiczne leczenie złamań i/lub deformacji kości;
- Opieka stomatologiczna w celu ochrony zdrowia zębów;
- Terapie przeciwbólowe w przypadku bardzo bolesnych złamań wielokrotnych;
- Fizjoterapia, do wydłużania i wzmacniania mięśni Elastyczny i tonizujący aparat mięśniowy pozwala zapobiegać upadkom, które mogą prowadzić do różnych złamań kości;
- Stosowanie środków ułatwiających poruszanie się, w tym wózków inwalidzkich, szelek, kul itp.
KORZYŚCI Z RUCHU
Osobom z wrodzoną łamliwością kości lekarze zalecają stałą praktykę ćwiczeń fizycznych i ogólnie ruchu, ponieważ obie te czynności przyczyniają się do wzmocnienia układu kostnego i mięśniowego.
Wśród zalecanych sportów są: pływanie, ponieważ jest to „aktywność fizyczna o niewielkim wpływie na układ kostny” oraz chodzenie.
KORZYŚCI ZE ZDROWEGO STYLU ŻYCIA
Prowadzenie zdrowego trybu życia, unikanie palenia, nadmierne spożywanie alkoholu, nadmierne i złe jedzenie itp. ma więcej niż dyskretne korzyści zdrowotne dla pacjentów z wrodzoną łamliwością kości, ponieważ spowalnia postęp choroby i zmniejsza kruchość kości.
ZABIEGI OBJAWOWE W FAZIE EKSPERYMENTALNEJ
Obecnie lekarze i badacze oceniają skuteczność niektórych terapii objawowych, w tym leczenia hormonem wzrostu oraz dożylnej i doustnej terapii opartej na bisfosfonianach.
Na chwilę obecną wyniki uzyskane dzięki wyżej wymienionym badaniom eksperymentalnym dobrze wróżą całemu środowisku medycznemu.
Rokowanie
Osteogenesis imperfecta jest chorobą o negatywnym rokowaniu, ponieważ jest nieuleczalna, drastycznie pogarsza jakość życia, aw niektórych przypadkach powoduje przedwczesną śmierć chorego.
Należy jednak zauważyć, że także dzięki nowoczesnym terapiom objawowym wiele osób z łagodną postacią wady wrodzonej osteogenezy jest w stanie prowadzić przyjemne i satysfakcjonujące życie.
Zapobieganie
Niestety nie ma obecnie środków zapobiegawczych przeciwko wrodzonej osteogenezie.


















.jpg)