Składniki aktywne: Ranibizumab
Lucentis 10 mg/ml roztwór do wstrzykiwań
Ulotki informacyjne Lucentis są dostępne dla wielkości opakowań:- Lucentis 10 mg/ml roztwór do wstrzykiwań
- Lucentis 10 mg/ml roztwór do wstrzykiwań w ampułko-strzykawce
Wskazania Dlaczego stosuje się Lucentis? Po co to jest?
Co to jest Lucentis?
Lucentis to roztwór, który należy wstrzykiwać do oka.Lucentis należy do grupy leków zwanych środkami przeciwnowotworowymi i zawiera substancję czynną o nazwie ranibizumab.
Do czego służy Lucentis
Lucentis stosuje się u osób dorosłych w leczeniu różnych chorób oczu powodujących pogorszenie widzenia.
Warunki te wynikają z uszkodzenia siatkówki (światłoczułej warstwy w tylnej części oka) spowodowanego:
- Wzrost nieprawidłowych naczyń krwionośnych, które wypuszczają płyny (neowaskularyzacja naczyniówki, CNV). Jest to widoczne w stanach takich jak związane z wiekiem zwyrodnienie plamki żółtej (AMD) lub patologiczna krótkowzroczność (PM).
- Obrzęk plamki (obrzęk w środkowej części siatkówki). Ten obrzęk może być spowodowany cukrzycą (stan zwany cukrzycowym obrzękiem plamki żółtej (DME)) lub zablokowaniem żył siatkówki (stan zwany niedrożnością żył siatkówki (RVO)).
Jak działa Lucentis
Lucentis specyficznie rozpoznaje i wiąże obecne w oku białko zwane ludzkim śródbłonkowym czynnikiem wzrostu naczyń A (VEGF-A). wzroku w warunkach takich jak AMD, PM, DME lub RVO. Wiążąc VEGF-A, Lucentis może blokować jego działanie i zapobiegać nieprawidłowemu wzrostowi i obrzękowi.
W takich warunkach Lucentis może pomóc ustabilizować, aw wielu przypadkach poprawić widzenie.
Przeciwwskazania Kiedy nie należy stosować produktu Lucentis
Nie możesz otrzymywać Lucentis
- jeśli pacjent ma uczulenie na ranibizumab lub którykolwiek z pozostałych składników tego leku
- jeśli masz „infekcję w jednym oku lub okolicy.
- jeśli u pacjenta występuje ból lub zaczerwienienie (ciężkie zapalenie wewnątrzgałkowe) w jednym oku.
Środki ostrożności dotyczące stosowania Informacje ważne przed przyjęciem leku Lucentis
Przed otrzymaniem leku Lucentis należy porozmawiać z lekarzem.
- Lucentis podaje się jako „wstrzyknięcie” do oka. Czasami może wystąpić „infekcja w wewnętrznej części oka”, ból lub zaczerwienienie (stan zapalny) odwarstwienie lub pęknięcie jednej z warstw w tylnej części oka (odwarstwienie lub pęknięcie siatkówki i odwarstwienie lub pęknięcie nabłonka) leczenie barwnikiem siatkówki Lucentis) lub zmętnienie soczewki (zaćma). Ważne jest, aby jak najszybciej zidentyfikować i leczyć „zakażenie lub odwarstwienie siatkówki. Należy natychmiast poinformować lekarza, jeśli wystąpią jakiekolwiek objawy, takie jak ból oka lub zwiększony dyskomfort, pogorszenie efektu czerwonych oczu”, niewyraźne lub osłabione widzenie, zwiększenie liczby krwinek w widzeniu lub zwiększonej wrażliwości na światło.
- U niektórych pacjentów ciśnienie w oku może na krótko wzrosnąć bezpośrednio po wstrzyknięciu. To zdarzenie jest czymś, czego możesz nie zauważyć, dlatego lekarz powinien sprawdzać po każdym wstrzyknięciu.
- Należy poinformować lekarza, jeśli pacjent miał w przeszłości problemy z oczami lub leczenie, lub wystąpił udar mózgu lub objawy przemijających ataków niedokrwiennych (osłabienie lub paraliż kończyn lub twarzy, trudności w mówieniu lub rozumieniu). Informacje te zostaną wzięte pod uwagę podczas oceny, czy lek Lucentis jest odpowiednim leczeniem dla Ciebie.
Dzieci i młodzież (poniżej 18 roku życia)
Stosowanie leku Lucentis u dzieci i młodzieży nie było badane i dlatego nie jest zalecane.
Interakcje Jakie leki lub pokarmy mogą zmienić działanie leku Lucentis
Należy poinformować lekarza, jeśli pacjent stosuje, ostatnio stosował lub może stosować jakiekolwiek inne leki.
Ostrzeżenia Ważne jest, aby wiedzieć, że:
Ciąża i karmienie piersią
- Kobietom w wieku rozrodczym należy zalecić stosowanie skutecznej antykoncepcji podczas leczenia.
- Nie ma doświadczenia ze stosowaniem leku Lucentis u kobiet w ciąży, dlatego potencjalne zagrożenia nie są znane.Jeśli pacjentka jest w ciąży, podejrzewa, że może być w ciąży lub planuje zajść w ciążę, powinna omówić to z lekarzem przed rozpoczęciem leczenia lekiem Lucentis.
- Nie zaleca się stosowania produktu Lucentis podczas karmienia piersią, ponieważ nie wiadomo, czy Lucentis przenika do mleka ludzkiego. Przed rozpoczęciem leczenia lekiem Lucentis należy zasięgnąć porady lekarza lub farmaceuty.
Prowadzenie i używanie maszyn
Po leczeniu produktem Lucentis może wystąpić przejściowe niewyraźne widzenie. W takim przypadku nie należy prowadzić pojazdów ani obsługiwać maszyn do czasu ustąpienia tego stanu.
Dawka, metoda i czas podawania Jak stosować lek Lucentis: dawkowanie
W jaki sposób otrzymacie Lucentis
Lek Lucentis jest podawany przez lekarza okulistę w postaci pojedynczego wstrzyknięcia do oka w znieczuleniu miejscowym.Zwykła dawka jednego wstrzyknięcia wynosi 0,05 ml (co zawiera 0,5 mg substancji czynnej). Odstęp między dwiema dawkami wstrzykniętymi do tego samego oka powinien wynosić co najmniej cztery tygodnie.Wszystkie wstrzyknięcia zostaną podane przez lekarza okulistę.
Przed wstrzyknięciem lekarz dokładnie oczyści oko, aby zapobiec infekcji. Lekarz poda również znieczulenie miejscowe, aby zmniejszyć lub zapobiec bólowi, który może wystąpić podczas wstrzyknięcia.
Leczenie rozpoczęto od jednego wstrzyknięcia leku Lucentis na miesiąc. Lekarz będzie monitorował stan oka i na podstawie odpowiedzi na leczenie zdecyduje, czy i kiedy konieczne jest dalsze leczenie.
Szczegółowe instrukcje dla użytkownika można znaleźć na końcu tej ulotki w punkcie „Jak przygotować i podawać lek Lucentis”.
Pacjenci w podeszłym wieku (65 lat i starsi)
Lucentis można stosować u pacjentów w wieku 65 lat i starszych bez dostosowania dawki.
Pominięcie dawki leku Lucentis
Jak najszybciej skontaktuj się z lekarzem lub szpitalem, aby przełożyć wizytę.
Przed przerwaniem leczenia produktem Lucentis
Jeśli rozważają Państwo przerwanie leczenia lekiem Lucentis, należy udać się na następną wizytę i omówić to z lekarzem. Lekarz doradzi i zadecyduje, jak długo będzie konieczne leczenie lekiem Lucentis.
W przypadku dalszych pytań dotyczących stosowania tego leku należy zwrócić się do lekarza.
Skutki uboczne Jakie są skutki uboczne Lucentis?
Jak każdy lek, lek ten może powodować działania niepożądane, chociaż nie u każdego one wystąpią.
Działania niepożądane związane z podawaniem leku Lucentis są spowodowane zarówno samym lekiem, jak i procedurą wstrzyknięcia i dotyczą głównie oka.
Najpoważniejsze skutki uboczne opisano poniżej:
Częste ciężkie działania niepożądane (mogą wystąpić u nie więcej niż 1 na 10 pacjentów): odwarstwienie lub łzawienie tylnej części oka (odwarstwienie lub pęknięcie siatkówki), objawiające się błyskami światła z mętami, aż do przejściowego pogorszenia widzenia lub zmętnienia soczewki (zaćma) Niezbyt częste poważne działania niepożądane (mogą wystąpić u nie więcej niż 1 na 100 pacjentów): ślepota, zakażenie gałki ocznej (zapalenie wnętrza gałki ocznej) ze stanem zapalnym wewnątrz oka.
Objawy, które mogą wystąpić, zostały opisane w punkcie 2 tej ulotki (należy przeczytać punkt 2 „Informacje ważne przed zastosowaniem leku Lucentis”). W przypadku wystąpienia któregokolwiek z tych działań niepożądanych należy natychmiast skontaktować się z lekarzem.
Poniżej opisano najczęściej zgłaszane działania niepożądane:
Bardzo częste działania niepożądane (mogą wystąpić u więcej niż 1 na 10 pacjentów)
Wizualne działania niepożądane obejmują: zapalenie oka, krwotok w tylnej części oka (krwotok siatkówkowy), zaburzenia widzenia, ból oka, cząstki lub plamki w polu widzenia (mroczki), miejscowe zaczerwienienie oka, podrażnienie oka, uczucie ciała obce oko, zwiększone wytwarzanie łez, zapalenie lub zakażenie brzegów powiek, suchość oka, zaczerwienienie lub swędzenie oka i zwiększone ciśnienie w oku.
Niewizualne skutki uboczne obejmują: ból gardła, przekrwienie błony śluzowej nosa, katar, ból głowy i ból stawów.
Poniżej opisano inne działania niepożądane, które mogą wystąpić po leczeniu lekiem Lucentis:
Częste skutki uboczne
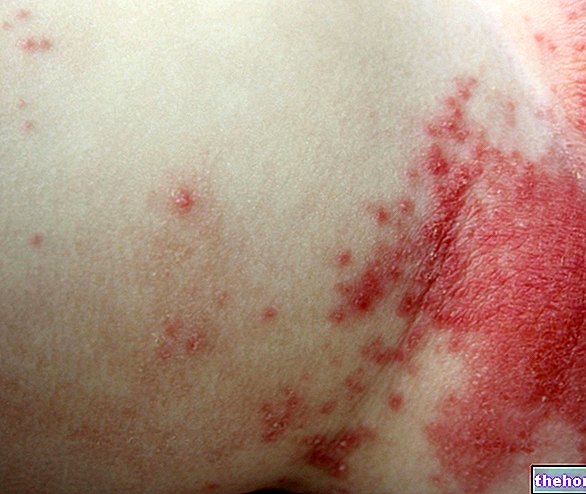
Działania niepożądane dotyczące wzroku obejmują: zmniejszoną ostrość widzenia, obrzęk części oka (błona naczyniowego, rogówki), zapalenie rogówki (przód oka), drobne plamki na powierzchni oka, niewyraźne widzenie, krwawienie w miejscu wstrzyknięcia, krwawienie w oku, swędząca wydzielina z oka, zaczerwienienie i obrzęk (zapalenie spojówek), nadwrażliwość na światło, dyskomfort w oku, obrzęk powiek, ból powiek Niewizualne działania niepożądane obejmują: zakażenie dróg moczowych, zmniejszenie liczby czerwonych krwinek (z objawami takimi jak: takie jak zmęczenie, duszność, zawroty głowy, bladość), lęk, kaszel, nudności, reakcje alergiczne, takie jak wysypka, pokrzywka, swędzenie i zaczerwienienie skóry.
Niezbyt częste skutki uboczne
Wizualne skutki uboczne obejmują: zapalenie i krwawienie w przedniej części oka, gromadzenie się ropy w oku, zmiany w centralnej części powierzchni oka, ból lub podrażnienie w miejscu wstrzyknięcia, nieprawidłowe czucie w oku, podrażnienie powiek.
Jeśli wystąpią jakiekolwiek działania niepożądane, należy porozmawiać z lekarzem, w tym wszelkie możliwe działania niepożądane niewymienione w tej ulotce.
Zgłaszanie skutków ubocznych
Jeśli wystąpią jakiekolwiek działania niepożądane, należy porozmawiać z lekarzem, w tym wszelkie możliwe działania niepożądane niewymienione w tej ulotce. Działania niepożądane można również zgłaszać bezpośrednio za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V. Zgłaszanie działań niepożądanych może pomóc w uzyskaniu większej ilości informacji na temat bezpieczeństwa tego leku.
Wygaśnięcie i przechowywanie
- Lek należy przechowywać w miejscu niewidocznym i niedostępnym dla dzieci.
- Nie stosować tego leku po upływie terminu ważności zamieszczonego na pudełku i etykiecie fiolki po EXP i EXP.Termin ważności oznacza ostatni dzień podanego miesiąca.
- Przechowywać w lodówce (2?C - 8?C). Nie zamrażać.
- Fiolkę przechowywać w opakowaniu zewnętrznym w celu ochrony leku przed światłem.
- Nie używaj uszkodzonego opakowania.
Zawartość opakowania i inne informacje
Co zawiera lek Lucentis
- Substancją czynną jest ranibizumab (10 mg/ml). Każdy ml zawiera 10 mg ranibizumabu.
- Pozostałe składniki to dwuwodzian α,α-trehalozy; chlorowodorek histydyny, monohydrat; histydyna; polisorbat 20; woda do wstrzykiwań.
Opis wyglądu leku Lucentis i co zawiera opakowanie
Lucentis to roztwór do wstrzykiwań w fiolce (0,23 ml). Roztwór jest wodny, klarowny, bezbarwny do jasnożółtego.
Lucentis jest dostarczany w opakowaniu zawierającym szklaną fiolkę z ranibizumabem z korkiem z gumy chlorobutylowej, tępą igłę z filtrem (18G x 1½″, 1,2 mm x 40 mm, 5 mikrometrów) do pobrania zawartości fiolki, igłę do wstrzykiwań (30G x ½ ", 0,3 mm x 13 mm) i strzykawkę (1 ml) do pobrania zawartości fiolki do wstrzyknięcia do ciała szklistego. Wszystkie składniki są przeznaczone wyłącznie do jednorazowego użytku.
Poniższe informacje są przeznaczone wyłącznie dla fachowego personelu medycznego: Należy również zapoznać się z rozdziałem „W jaki sposób lek Lucentis zostanie podany pacjentowi”.
Jak przygotować i podawać Lucentis
Fiolki jednorazowego użytku przeznaczone wyłącznie do podania do ciała szklistego Produkt Lucentis powinien być podawany przez wykwalifikowanego okulistę, posiadającego doświadczenie w wykonywaniu wstrzyknięć do ciała szklistego.
W wysiękowym AMD, zaburzeniach widzenia spowodowanych DME, obrzęku plamki wtórnym do RVO lub CNV wtórnym do PM, zalecana dawka produktu Lucentis wynosi 0,5 mg w pojedynczym wstrzyknięciu do ciała szklistego.
Odpowiada to wstrzykniętej objętości 0,05 ml. Odstęp między dwiema dawkami wstrzykniętymi do tego samego oka powinien wynosić co najmniej cztery tygodnie.
Leczenie rozpoczyna się od jednego wstrzyknięcia na miesiąc, aż do uzyskania maksymalnej ostrości wzroku i (lub) braku oznak aktywności choroby, takich jak zmiany ostrości wzroku i zmiany innych objawów przedmiotowych i podmiotowych choroby podczas kontynuowania leczenia pacjenci z wysiękową postacią AMD, DME i RVO, może być konieczne rozpoczęcie terapii trzema lub więcej kolejnymi wstrzyknięciami co miesiąc.
Dlatego odstępy monitorowania i leczenia powinny być ustalane przez lekarza i powinny być oparte na aktywności choroby, co potwierdza ocena ostrości wzroku i/lub parametrów anatomicznych.
Jeżeli w opinii lekarza ostrość wzroku i parametry anatomiczne wskazują, że pacjent nie odnosi korzyści z dalszego leczenia, produkt Lucentis należy przerwać.
Monitorowanie aktywności choroby może obejmować badanie kliniczne, ocenę czynnościową lub techniki obrazowania (takie jak optyczna koherentna tomografia lub angiografia fluoresceinowa).
Jeśli pacjenci są leczeni zgodnie ze schematem leczenia i wydłużania, po osiągnięciu maksymalnej ostrości wzroku i (lub) przy braku oznak aktywności choroby, odstępy leczenia można stopniowo wydłużać do wystąpienia objawów choroby lub pogorszenia widzenia. funkcjonować. Odstęp leczenia powinien być stopniowo wydłużany o maksymalnie dwa tygodnie u pacjentów z wysiękową postacią AMD i może być wydłużony do jednego miesiąca u pacjentów z DME.Odstępy między zabiegami można również stopniowo wydłużać w leczeniu RVO, jednak nie są wystarczające dane do ustalenia czasu trwania tych interwałów. W przypadku ponownego wystąpienia choroby należy odpowiednio skrócić przerwę w leczeniu.
W leczeniu zaburzeń widzenia spowodowanych CNV wtórną do PM wielu pacjentów wymaga tylko jednego lub dwóch wstrzyknięć w ciągu pierwszego roku, a niektórzy wymagają częstszego leczenia.
Lucentis i fotokoagulacja laserowa w DME i obrzęku plamki wtórnym do BRVO
Istnieją pewne doświadczenia związane ze stosowaniem produktu Lucentis jednocześnie z fotokoagulacją laserową.W przypadku stosowania tego samego dnia, produkt Lucentis należy podać co najmniej 30 minut po fotokoagulacji laserowej.Lucentis można podawać pacjentom, którzy przeszli wcześniej fotokoagulację laserową.
Lucentis i terapia fotodynamiczna z Visudyne w CNV wtórnej do PM
Brak doświadczenia w podawaniu produktu Lucentis w skojarzeniu z Visudyne. Przed podaniem produkt Lucentis należy sprawdzić wzrokowo pod kątem obecności cząstek i przebarwień.
Zabieg wstrzyknięcia powinien być wykonywany w warunkach aseptycznych, które obejmują chirurgiczną dezynfekcję rąk, sterylne rękawiczki, sterylną obłożenie i sterylny blefarostat (lub odpowiednik) oraz możliwość wykonania sterylnej paracentezy (w razie potrzeby). wywiad lekarski należy dokładnie zbadać pod kątem reakcji nadwrażliwości. Zgodnie z praktyką kliniczną przed wstrzyknięciem należy zastosować odpowiednie znieczulenie i zastosować miejscowo środek przeciwdrobnoustrojowy o szerokim spektrum w celu dezynfekcji powierzchni okołogałkowej, oka i powiek.
Aby przygotować lek Lucentis do wstrzyknięcia do ciała szklistego, należy postępować zgodnie z poniższymi instrukcjami:
Wszystkie składniki są sterylne i przeznaczone wyłącznie do jednorazowego użytku. Nie wolno używać żadnego elementu, którego opakowanie nosi ślady uszkodzenia lub manipulacji. Nie można zagwarantować sterylności, jeśli uszczelnienie opakowania komponentu nie jest nienaruszone.
- Zdezynfekować zewnętrzną część gumowego korka fiolki przed pobraniem.
- Aseptycznie przymocuj igłę z filtrem 5 µm (18G x 1½″, 1,2 mm x 40 mm, 5 µm, w zestawie) do 1 ml strzykawki (w zestawie). Włóż tępą igłę z filtrem do środka nasadki, aż dotknie dna fiolkę.
- Pobrać cały płyn z fiolki, trzymając ją w pozycji pionowej, lekko pochylonej, aby ułatwić całkowite pobranie.
- Upewnić się, że tłok strzykawki jest odciągnięty wystarczająco daleko podczas opróżniania fiolki, aby całkowicie opróżnić igłę z filtrem.
- Igłę z filtrem pozostawić stępioną w fiolce i wyjąć z niej strzykawkę.Po pobraniu zawartości fiolki igłę z filtrem należy wyrzucić i nie używać jej do wstrzyknięcia do ciała szklistego.
- Bezpiecznie i aseptycznie zamocuj igłę iniekcyjną (30G x ½ ″, 0,3 mm x 13 mm, w zestawie) na strzykawce.
- Ostrożnie zdjąć nasadkę z igły iniekcyjnej bez odłączania igły iniekcyjnej od strzykawki. Uwaga: Przytrzymaj żółtą podstawę igły do wstrzykiwań podczas zdejmowania nasadki.
- Ostrożnie usunąć powietrze ze strzykawki i dostosować dawkę do 0,05 ml zaznaczonej na strzykawce Strzykawka jest gotowa do wstrzyknięcia. Uwaga: Nie czyścić igły do wstrzyknięć. Nie odciągać tłoka do tyłu. Wprowadzić igłę do wstrzyknięć 3,5-4,0 mm za rąbkiem do komory ciała szklistego, unikając południka poziomego i kierując igłę w kierunku środka kuli. objętość wtrysku 0,05 ml; zmienić miejsce na twardówce dla kolejnych wstrzyknięć. Po wstrzyknięciu nie zakrywać igły ani nie odłączać jej od strzykawki. Zużytą strzykawkę wraz z igłą wyrzucić do odpowiedniego pojemnika lub zgodnie z lokalnymi przepisami.
Ulotka pakietu źródłowego: AIFA (Włoska Agencja Leków). Treść opublikowana w styczniu 2016 r. Przedstawione informacje mogą być nieaktualne.
Aby mieć dostęp do najbardziej aktualnej wersji, warto wejść na stronę AIFA (Włoskiej Agencji Leków). Zastrzeżenie i przydatne informacje.
01.0 NAZWA PRODUKTU LECZNICZEGO
LUCENTIS 10 MG/ML ROZTWÓR DO WSTRZYKIWAŃ
02.0 SKŁAD JAKOŚCIOWY I ILOŚCIOWY
Jeden ml zawiera 10 mg ranibizumabu*. Każda fiolka zawiera 2,3 mg ranibizumabu w 0,23 ml roztworu.
* Ranibizumab jest fragmentem humanizowanego przeciwciała monoklonalnego wytwarzanym w komórkach Escherichia coli za pomocą technologii rekombinacji DNA.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
03.0 POSTAĆ FARMACEUTYCZNA
Roztwór do wstrzykiwań
Przejrzysty, bezbarwny do jasnożółtego roztwór wodny.
04.0 INFORMACJE KLINICZNE
04.1 Wskazania terapeutyczne
Lucentis jest wskazany u dorosłych w:
• Leczenie związanego z wiekiem neowaskularnego (mokrego) zwyrodnienia plamki żółtej (AMD)
• Leczenie zaburzeń widzenia spowodowanych cukrzycowym obrzękiem plamki (DME)
• Leczenie zaburzeń widzenia spowodowanych „obrzękiem plamki żółtej wtórnym do niedrożności żył siatkówki (RVO rozgałęzione lub RVO centralne)
• Leczenie zaburzeń widzenia spowodowanych neowaskularyzacją naczyniówki (CNV) wtórną do krótkowzroczności patologicznej (PM)
04.2 Dawkowanie i sposób podawania
Produkt Lucentis powinien być podawany przez wykwalifikowanego okulistę, posiadającego doświadczenie w wykonywaniu wstrzyknięć do ciała szklistego.
Dawkowanie w leczeniu wysiękowego AMD
Zalecana dawka produktu Lucentis to 0,5 mg podawane co miesiąc w pojedynczym wstrzyknięciu do ciała szklistego. Odpowiada to wstrzykniętej objętości 0,05 ml.
Leczenie podaje się co miesiąc i kontynuuje aż do uzyskania maksymalnej ostrości wzroku, tj. ostrości wzroku pacjenta ustabilizuje się w trzech kolejnych comiesięcznych kontrolach wykonywanych podczas leczenia ranibizumabem.
Dlatego ostrość wzroku pacjentów powinna być monitorowana co miesiąc.
Leczenie należy wznowić, gdy monitorowanie wskazuje na pogorszenie ostrości wzroku spowodowane wysiękową postacią AMD.Następnie comiesięczne wstrzyknięcia należy podawać aż do uzyskania stabilnej ostrości wzroku przez trzy kolejne comiesięczne kontrole (co oznacza co najmniej dwa wstrzyknięcia). Odstęp między dwiema dawkami nie powinien być krótszy niż jeden miesiąc.
Dawkowanie w leczeniu zaburzeń widzenia spowodowanych przez DME lub obrzęk plamki wtórny do RVO
Zalecana dawka produktu Lucentis to 0,5 mg podawane co miesiąc w pojedynczym wstrzyknięciu do ciała szklistego. Odpowiada to wstrzykniętej objętości 0,05 ml.
Leczenie podaje się co miesiąc i kontynuuje aż do uzyskania maksymalnej ostrości wzroku, tj. ostrości wzroku pacjenta ustabilizuje się w trzech kolejnych comiesięcznych kontrolach wykonywanych podczas leczenia ranibizumabem. Jeśli nie ma poprawy ostrości wzroku w okresie pierwszych trzech wstrzyknięć, nie zaleca się kontynuacji leczenia.
Dlatego ostrość wzroku pacjentów powinna być monitorowana co miesiąc.
Leczenie należy wznowić, gdy monitorowanie wykaże pogorszenie ostrości wzroku spowodowane DME lub obrzękiem plamki wtórnym do RVO.Następnie comiesięczne wstrzyknięcia należy podawać aż do uzyskania stabilnej ostrości wzroku przez trzy kolejne comiesięczne kontrole (co wymaga co najmniej dwóch wstrzyknięć). Odstęp między dwiema dawkami nie powinien być krótszy niż jeden miesiąc.
Lucentis i fotokoagulacja laserowa w DME i obrzęku plamki wtórnym do BRVO
Istnieją pewne doświadczenia dotyczące podawania produktu Lucentis jednocześnie z fotokoagulacją laserową (patrz punkt 5.1). W przypadku podania tego samego dnia, produkt Lucentis należy podać co najmniej 30 minut po fotokoagulacji laserowej. Lucentis można podawać pacjentom, którzy wcześniej przeszli fotokoagulację laserową.
Dawkowanie w leczeniu zaburzeń widzenia spowodowanych CNV wtórną do PM
Leczenie należy rozpocząć od pojedynczego wstrzyknięcia.
Jeśli monitorowanie wykaże oznaki aktywności choroby, takie jak obniżona ostrość wzroku i (lub) oznaki urazu, zalecane jest dalsze leczenie.
Monitorowanie choroby może obejmować badanie kliniczne, optyczną koherentną tomografię (OCT) lub angiografię fluoresceinową (FA).
Chociaż niektórzy pacjenci mogą potrzebować tylko jednego lub dwóch wstrzyknięć w pierwszym roku leczenia, niektórzy mogą wymagać częstszego leczenia (patrz punkt 5.1). Dlatego zaleca się comiesięczne monitorowanie przez pierwsze dwa miesiące i co najmniej co trzy miesiące przez pierwszy rok leczenia. Po pierwszym roku częstotliwość monitorowania może ustalić lekarz.
Odstęp między dwiema dawkami nie powinien być krótszy niż jeden miesiąc.
Lucentis i terapia fotodynamiczna z Visudyne w CNV wtórnej do PM
Brak doświadczenia w podawaniu produktu Lucentis w skojarzeniu z Visudyne.
Populacje specjalne
Niewydolność wątroby
Produktu Lucentis nie badano u pacjentów z niewydolnością wątroby. Jednak nie są potrzebne żadne specjalne względy dla tego polowania.
Niewydolność nerek
Nie ma konieczności dostosowania dawki u pacjentów z niewydolnością nerek (patrz punkt 5.2).
Starsi mieszkańcy
Nie ma konieczności dostosowania dawki u osób w podeszłym wieku. Doświadczenie dotyczące pacjentów z DME w wieku powyżej 75 lat jest „ograniczone”.
Populacja pediatryczna
Nie ustalono bezpieczeństwa i skuteczności stosowania produktu Lucentis u dzieci i młodzieży w wieku poniżej 18 lat Brak dostępnych danych.
Sposób podawania
Fiolki jednorazowego użytku wyłącznie do podania do ciała szklistego.
Przed podaniem produkt Lucentis należy sprawdzić wzrokowo pod kątem obecności cząstek i przebarwień.
Zabieg wstrzyknięcia musi być wykonywany w warunkach aseptycznych, które obejmują dezynfekcję rąk jak w przypadku każdego zabiegu chirurgicznego, sterylne rękawiczki, sterylną chusteczkę i sterylny blefarostat (lub odpowiednik) oraz możliwość wykonania sterylnej paracentezy (w razie potrzeby). przed zabiegiem do ciała szklistego należy dokładnie ocenić historię reakcji nadwrażliwości u pacjenta (patrz punkt 4.4). Zgodnie z praktyką kliniczną przed wstrzyknięciem należy zastosować odpowiednie znieczulenie i zastosować miejscowo środek przeciwdrobnoustrojowy o szerokim spektrum w celu dezynfekcji powierzchni okołogałkowej, oka i powiek.
Informacje dotyczące przygotowania produktu Lucentis, patrz punkt 6.6.
Wprowadzić igłę iniekcyjną 3,5-4,0 mm za rąbkiem, do komory ciała szklistego, unikając południka poziomego i kierując igłę w kierunku środka gałki ocznej. Wstrzyknąć objętość wstrzyknięcia 0,05 ml; zmienić miejsce na twardówce przy kolejnych wstrzyknięciach.
04.3 Przeciwwskazania
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Pacjenci z aktualnymi lub podejrzewanymi infekcjami oka lub okołogałka.
Pacjenci z trwającym ciężkim zapaleniem wewnątrzgałkowym.
04.4 Specjalne ostrzeżenia i odpowiednie środki ostrożności dotyczące stosowania
Reakcje związane z wstrzyknięciem do ciała szklistego
Wstrzyknięcia do ciała szklistego, w tym wstrzyknięcia produktu Lucentis, były związane z zapaleniem wnętrza gałki ocznej, przedarciowym odwarstwieniem siatkówki, pęknięciem siatkówki i jatrogenną zaćmą urazową (patrz punkt 4.8). Do podawania produktu Lucentis należy zawsze stosować odpowiednie techniki aseptycznego wstrzykiwania. Ponadto pacjentów należy monitorować w ciągu tygodnia po wstrzyknięciu, aby umożliwić szybkie leczenie w przypadku infekcji. Pacjentów należy poinstruować, jak bezzwłocznie zgłaszać wszelkie objawy sugerujące zapalenie wnętrza gałki ocznej lub dowolne z powyższych zdarzeń.
Wzrost ciśnienia wewnątrzgałkowego
Przejściowy wzrost ciśnienia wewnątrzgałkowego (ang. intraocular pressure, IOP) obserwowano w ciągu 60 minut po wstrzyknięciu produktu Lucentis. Obserwowano również przedłużone podwyższenie ciśnienia wewnątrzgałkowego (patrz punkt 4.8).Należy monitorować i odpowiednio leczyć ciśnienie wewnątrzgałkowe i perfuzję w głowie nerwu wzrokowego.
Leczenie dwustronne
Ograniczone dane dotyczące dwustronnego stosowania produktu Lucentis (w tym dawkowania tego samego dnia) nie wskazują na zwiększone ryzyko ogólnoustrojowych zdarzeń niepożądanych w porównaniu z leczeniem jednostronnym.
Immunogenność
Istnieje możliwość immunogenności produktu Lucentis. Ponieważ istnieje możliwość zwiększonej ekspozycji ogólnoustrojowej u pacjentów z DME, nie można wykluczyć zwiększonego ryzyka rozwoju nadwrażliwości w tej populacji pacjentów.Pacjenci powinni być również poinformowani o tym, jak zgłaszać nasilenie zapalenia wewnątrzgałkowego, ponieważ może to być objaw kliniczny przypisywany tworzenie przeciwciał wewnątrzgałkowych.
Jednoczesne stosowanie z innymi czynnikami anty-VEGF (czynnik wzrostu śródbłonka naczyniowego)
Produktu Lucentis nie wolno podawać jednocześnie z innymi produktami leczniczymi anty-VEGF (ogólnie lub do oka).
Odstawienie Lucentisa
Nie należy podawać dawki i nie należy wznawiać leczenia przed następnym planowym leczeniem w przypadku:
• spadek w najlepszej skorygowanej ostrości wzroku (BCVA) ≥30 liter w porównaniu z ostatnią oceną;
• ciśnienie wewnątrzgałkowe ≥30 mmHg;
• pęknięcie siatkówki;
• „krwotok podsiatkówkowy rozciągający się do środka dołka lub jeśli krwotok ma zasięg ≥50% całkowitej powierzchni zmiany”;
• operacja wewnątrzgałkowa wykonana lub planowana w ciągu poprzednich lub następnych 28 dni.
Pęknięcie nabłonka barwnikowego siatkówki
Czynniki ryzyka związane z początkiem pęknięcia nabłonka barwnikowego siatkówki po terapii anty-VEGF w wysiękowej postaci AMD obejmują duże i/lub silne odwarstwienie nabłonka barwnikowego siatkówki.Rozpoczynając leczenie produktem Lucentis, należy zachować ostrożność u pacjentów z tymi czynnikami ryzyka pęknięcia nabłonka barwnikowego siatkówki.
Regmatogenne odwarstwienie siatkówki lub dziury w plamce
Leczenie należy przerwać u osób z przedarciowym odwarstwieniem siatkówki lub otworami w plamce 3. lub 4. stopnia.
Populacje z ograniczonymi danymi
Doświadczenie w leczeniu pacjentów z DME wtórnym do cukrzycy typu I jest ograniczone. . Lekarz powinien wziąć pod uwagę brak informacji podczas leczenia tych pacjentów.
U pacjentów z PM dostępne są ograniczone dane dotyczące wpływu produktu Lucentis na pacjentów wcześniej leczonych nieskuteczną terapią fotodynamiczną werteporfiną (vPDT).Ponadto, chociaż stały efekt obserwowano u pacjentów ze zmianami poddołkowymi i okołodołkowymi, nie ma wystarczających danych na temat wpływ produktu Lucentis u pacjentów z PM ze zmianami pozadołkowymi.
Działania ogólnoustrojowe po podaniu do ciała szklistego
Po wstrzyknięciu do ciała szklistego inhibitorów VEGF zgłaszano ogólnoustrojowe zdarzenia niepożądane, w tym krwotoki pozagałkowe i tętnicze zdarzenia zakrzepowo-zatorowe.
Istnieją ograniczone dane dotyczące bezpieczeństwa leczenia DME, obrzęku plamki wywołanego przez RVO i CNV wtórną do PM u pacjentów z udarem lub przemijającymi napadami niedokrwiennymi w wywiadzie.Należy zachować szczególną ostrożność podczas leczenia takich pacjentów (patrz punkt 4.8).
Poprzednie odcinki RVO, gałąź niedokrwienna i centralna RVO
Doświadczenie w leczeniu pacjentów z przebytymi epizodami RVO oraz pacjentów z niedokrwienną gałęzią RVO (BRVO) i centralnym RVO (CRVO) jest ograniczone. nie jest zalecane.
04.5 Interakcje z innymi produktami leczniczymi i inne formy interakcji
Nie przeprowadzono konwencjonalnych badań interakcji.
Informacje na temat skojarzonego stosowania terapii fotodynamicznej (PDT) z werteporfiną i produktem Lucentis w wysiękowej postaci AMD i PM, patrz punkt 5.1.
Informacje na temat skojarzonego stosowania fotokoagulacji laserowej i produktu Lucentis w leczeniu DME i BRVO, patrz punkty 4.2 i 5.1.
04.6 Ciąża i laktacja
Kobiety w wieku rozrodczym / antykoncepcja u kobiet
Kobiety w wieku rozrodczym powinny podczas leczenia stosować skuteczną antykoncepcję.
Ciąża
W przypadku ranibizumabu nie są dostępne żadne dane kliniczne dotyczące ekspozycji ciąż. Badania na małpach cynomolgus nie wykazały bezpośredniego ani pośredniego szkodliwego wpływu na ciążę lub rozwój zarodka/płodu (patrz punkt 5.3). Ekspozycja ogólnoustrojowa na ranibizumab po podaniu do oka jest niewielka, ale ze względu na mechanizm działania ranibizumab należy uznać za potencjalnie teratogenny i toksyczny dla zarodka/płodu. Dlatego ranibizumabu nie należy stosować w okresie ciąży, chyba że oczekiwane korzyści przewyższają potencjalne ryzyko dla płodu. Kobietom planującym zajście w ciążę i leczonym ranibizumabem zaleca się odczekanie co najmniej 3 miesięcy od ostatniej dawki ranibizumabu przed poczęciem dziecka.
Ciąża
Nie wiadomo, czy Lucentis przenika do mleka ludzkiego. Zaleca się, aby nie karmić piersią podczas stosowania leku Lucentis.
Płodność
Brak dostępnych danych dotyczących płodności.
04.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Procedura leczenia produktem Lucentis może wywołać przemijające zaburzenia widzenia, które mogą wpływać na zdolność prowadzenia pojazdów lub obsługiwania maszyn (patrz punkt 4.8). Pacjenci doświadczający tych objawów nie powinni prowadzić pojazdów ani obsługiwać maszyn do czasu ustąpienia tych przemijających zaburzeń widzenia.
04.8 Działania niepożądane
Podsumowanie profilu bezpieczeństwa
Większość działań niepożądanych zgłaszanych po podaniu produktu Lucentis jest związanych z zabiegiem wstrzyknięcia do ciała szklistego.
Najczęściej zgłaszanymi działaniami niepożądanymi dotyczącymi oka po wstrzyknięciu produktu Lucentis są: ból oka, przekrwienie oka, zwiększone ciśnienie śródgałkowe, zapalenie ciała szklistego, odwarstwienie ciała szklistego, krwotok siatkówkowy, zaburzenia widzenia, męty (męty w ciele szklistym), krwotok spojówkowy, podrażnienie oka, uczucie ciała obcego w oko, zwiększone łzawienie, zapalenie powiek, suchość i swędzenie oka.
Najczęściej zgłaszanymi działaniami niepożądanymi niezwiązanymi z oczami są bóle głowy, zapalenie nosogardzieli i bóle stawów.
Rzadziej zgłaszane, ale poważniejsze działania niepożądane obejmują zapalenie wnętrza gałki ocznej, ślepotę, odwarstwienie siatkówki, pęknięcie siatkówki i jatrogenną zaćmę pourazową (patrz punkt 4.4).
Pacjentów należy poinformować o objawach tych potencjalnych działań niepożądanych i poinstruować lekarza, aby poinformowali swojego lekarza, jeśli wystąpią takie objawy, jak ból oka lub zwiększony dyskomfort, nasilenie zaczerwienienia oka, niewyraźne lub osłabione widzenie, zwiększona liczba mętów w ciele szklistym lub „ zwiększona wrażliwość na światło.
Działania niepożądane zgłaszane po podaniu produktu Lucentis w badaniach klinicznych podsumowano w poniższej tabeli.
Tabela działań niepożądanych nr
Działania niepożądane wymieniono według klasyfikacji układów i narządów oraz częstości, stosując następującą konwencję: bardzo często (≥1/10), często (≥1/100,
Infekcje i infestacje
Bardzo częste Zapalenie nosogardzieli
pospolity Zakażenie dróg moczowych *
Zaburzenia układu krwionośnego i limfatycznego
pospolity Niedokrwistość
Zaburzenia układu odpornościowego
pospolity Nadwrażliwość
Zaburzenia psychiczne
pospolity Lęk
Zaburzenia układu nerwowego
Bardzo częste Bół głowy
Zaburzenia oka
Bardzo częste Zapalenie ciała szklistego, odwarstwienie ciała szklistego, krwotok siatkówkowy, zaburzenia widzenia, ból oka, męty w ciele szklistym, krwotok spojówkowy, podrażnienie oka, uczucie ciała obcego w oku, nasilone łzawienie, zapalenie powiek, suchość oka, przekrwienie oka, swędzenie oka.
pospolity Zwyrodnienie siatkówki, zaburzenia siatkówki, odwarstwienie siatkówki, rozerwanie siatkówki, odwarstwienie nabłonka barwnikowego siatkówki, rozerwanie nabłonka barwnikowego siatkówki, zaburzenia ostrości wzroku, krwotok do ciała szklistego, zaburzenia w ciele szklistym, zapalenie błony naczyniowej oka, zapalenie tęczówki, zapalenie tęczówki, zapalenie tęczówki, zaćma, zaćma podtorebkowa przednia zapalenie rogówki, otarcie rogówki, reakcja komory przedniej, niewyraźne widzenie, krwotok w miejscu wstrzyknięcia, krwotok do oka, zapalenie spojówek, zapalenie spojówek
uczulenie, wydzielina z oczu, błyski świetlne, światłowstręt, dyskomfort w oku, obrzęk powiek, ból powiek, przekrwienie spojówek.
Niezwykły Ślepota, zapalenie wnętrza gałki ocznej, hipopion, hyphema, keratopatia, zrosty tęczówki, złogi na rogówce, obrzęk rogówki, rozstępy rogówki, ból w miejscu wstrzyknięcia, podrażnienie w miejscu wstrzyknięcia, nieprawidłowe czucie w oku, podrażnienie powiek.
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia
pospolity Kaszel
Zaburzenia żołądkowo-jelitowe
pospolity Mdłości
Zaburzenia skóry i tkanki podskórnej
pospolity Reakcje alergiczne (wysypka, pokrzywka, swędzenie, rumień)
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej
Bardzo częste Ból stawów
Testy diagnostyczne
Bardzo częste Zwiększone ciśnienie wewnątrzgałkowe
# Działania niepożądane zdefiniowano jako zdarzenia niepożądane (u co najmniej 0,5 punktu procentowego pacjentów), które występowały częściej (co najmniej 2 punkty procentowe) u pacjentów otrzymujących produkt Lucentis 0,5 mg w porównaniu z tymi, którzy otrzymywali leczenie kontrolne (pozorowane lub PDT werteporfina).
* obserwowane tylko w populacji z DME
Działania niepożądane związane z kategorią leku
W badaniach fazy III wysiękowego AMD ogólna częstość krwotoków pozagałkowych, zdarzenia niepożądanego potencjalnie związanego z inhibitorami VEGF (czynnik wzrostu naczyń śródbłonka), była nieznacznie zwiększona u pacjentów leczonych ranibizumabem. wzór między różnymi krwotokami. Istnieje teoretyczne ryzyko wystąpienia tętniczych incydentów zakrzepowo-zatorowych, w tym udaru mózgu i zawału mięśnia sercowego, wynikające z doszklistkowego stosowania inhibitorów VEGF. W badaniach klinicznych produktu Lucentis u pacjentów z AMD, DME, RVO i PM obserwowano niewielką częstość występowania tętniczych zdarzeń zakrzepowo-zatorowych i nie zaobserwowano różnic między grupami ranibizumabu w porównaniu z grupą kontrolną.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie podejrzewanych działań niepożądanych, które występują po dopuszczeniu produktu leczniczego do obrotu, jest ważne, ponieważ umożliwia ciągłe monitorowanie stosunku korzyści do ryzyka produktu leczniczego. Pracownicy służby zdrowia proszeni są o zgłaszanie wszelkich podejrzewanych działań niepożądanych za pośrednictwem Włoskiej Agencji Leków. , strona internetowa: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Przedawkowanie
W badaniach klinicznych dotyczących wysiękowej postaci AMD i danych po wprowadzeniu produktu do obrotu zgłaszano przypadki przypadkowego przedawkowania.Działaniami niepożądanymi najczęściej związanymi z tymi przypadkami były zwiększone ciśnienie śródgałkowe, przemijająca ślepota, pogorszenie ostrości wzroku, obrzęk rogówki i ból. ciśnienie śródgałkowe powinno być monitorowane i leczone, jeśli lekarz uzna to za konieczne.
05.0 WŁAŚCIWOŚCI FARMAKOLOGICZNE
05.1 Właściwości farmakodynamiczne
Grupa farmakoterapeutyczna: leki okulistyczne, leki przeciwnowotworowe, kod ATC: S01LA04
Ranibizumab jest fragmentem humanizowanego rekombinowanego przeciwciała monoklonalnego skierowanego przeciwko ludzkiemu czynnikowi wzrostu śródbłonka naczyniowego typu A (VEGF-A). Wiąże się z wysokim powinowactwem do izoform VEGF-A (np. VEGF110, VEGF121 i VEGF165), zapobiegając w ten sposób wiązaniu VEGF-A z jego receptorami VEGFR-1 i VEGFR-2. neowaskularyzacja i wzrost przepuszczalności naczyń, które uważa się za przyczyniające się do progresji neowaskularnej postaci zwyrodnienia plamki żółtej związanego z wiekiem, krótkowzroczności patologicznej lub pogorszenia widzenia spowodowanego albo cukrzycowym obrzękiem plamki żółtej albo „obrzękiem plamki wtórnym do RVO.
Leczenie wysiękowego AMD
W przypadku wysiękowej postaci AMD bezpieczeństwo i skuteczność kliniczną produktu Lucentis oceniano w trzech 24-miesięcznych randomizowanych badaniach z podwójnie ślepą próbą, kontrolowanych terapią pozorowaną lub substancją czynną u pacjentów z wysiękową postacią AMD. Łącznie do tych badań włączono 1323 pacjentów (879 leczonych i 444 z grupy kontrolnej).
W badaniu FVF2598g (MARINA) 716 pacjentów z minimalnie klasycznymi lub utajonymi zmianami neowaskularyzacji naczyniówkowej (CNV) bez klasycznego komponentu otrzymywało comiesięczne wstrzyknięcia do ciała szklistego produktu Lucentis w dawce 0,3 mg (n = 238) lub 0,5 mg (n = 240) lub wstrzyknięcia pozorowane (n = 238).
W badaniu FVF2587g (ANCHOR) 423 pacjentów z głównie klasyczną CNV otrzymało jedną z następujących metod leczenia: 1) comiesięczne wstrzyknięcia do ciała szklistego produktu Lucentis 0,3 mg i PDT pozorowane (n = 140); 2) comiesięczne wstrzyknięcia do ciała szklistego produktu Lucentis 0,5 mg i pozorowane PDT (n = 140); lub 3) doszklistkowe iniekcje pozorowane i PDT z werteporfiną (n = 143). PDT z werteporfiną lub pozorowane podawano razem z początkowym wstrzyknięciem Lucentis, a następnie co 3 miesiące, jeśli fluorangiografia wykazała utrzymywanie się lub wznowienie przecieku naczyniowego.
Kluczowe ustalenia podsumowano w tabelach 1, 2 i na rysunku 1.
Tabela 1 Wyniki w 12. i 24. miesiącu w badaniu FVF2598g (MARINA)
ap
Tabela 2 Wyniki w 12. i 24. miesiącu badania FVF2587g (ANCHOR)
Tabela
Wyniki obu badań wykazały, że kontynuacja leczenia ranibizumabem może być również korzystna u pacjentów, którzy utracili ≥15 liter najlepszej skorygowanej ostrości wzroku (BCVA) w pierwszym roku leczenia.
Badanie FVF3192g (PIER) było randomizowanym, podwójnie ślepym, kontrolowanym badaniem pozorowanym, zaprojektowanym w celu oceny bezpieczeństwa i skuteczności produktu Lucentis u 184 pacjentów ze wszystkimi postaciami wysiękowej postaci AMD. 0,5 mg (n = 61) lub wstrzyknięcia pozorowane (n = 63) raz w miesiącu przez 3 kolejne dawki, a następnie jedną dawkę podawaną raz na 3 miesiące Od 14 miesiąca badania pacjenci leczeni wstrzyknięciem pozorowanym byli przyjmowani do leczenie ranibizumabem, a od 19. miesiąca można było wykonywać częstsze zabiegi. Pacjenci leczeni produktem Lucentis w badaniu PIER otrzymali łącznie średnio 10 zabiegów.
Pierwszorzędowym punktem końcowym skuteczności była średnia zmiana ostrości wzroku po 12 miesiącach w porównaniu ze stanem wyjściowym. Po początkowym wzroście ostrości wzroku (po dawce miesięcznej) ostrość wzroku pacjentów zmniejszała się średnio przy dawkowaniu kwartalnym, powracając do wartości wyjściowej w 12. miesiącu i efekt ten utrzymywał się u większości leczonych pacjentów. Miesiąc 24. Dane od ograniczonej liczby pacjentów, których przeniesiono na leczenie ranibizumabem po ponad roku leczenia pozorowanego, sugerowały, że wczesne rozpoczęcie leczenia może być związane z lepszym zachowaniem „ostrości wzroku”.
W obu badaniach MARINA i ANCHOR poprawie ostrości wzroku obserwowanej po zastosowaniu produktu Lucentis 0,5 mg po 12 miesiącach towarzyszyły korzyści zgłaszane przez pacjentów, mierzone za pomocą kwestionariusza National Eye Institute Visual Function Questionnaire (VFQ-25). a dwie grupy kontrolne zostały ocenione z wartościami p w zakresie od 0,009 do
Skuteczność produktu Lucentis w leczeniu wysiękowego AMD została potwierdzona w badaniach AMD po wprowadzeniu produktu do obrotu. Dane z dwóch badań (MONT BLANC, BPD952A2308 i DENALI, BPD952A2309) nie wykazały dodatkowych skutków. tylko do Lucentis.
Leczenie wad wzroku spowodowanych DME
Bezpieczeństwo i skuteczność produktu Lucentis oceniano w dwóch 12-miesięcznych randomizowanych badaniach z podwójnie ślepą próbą, z grupą kontrolną otrzymującą leczenie pozorowane lub z leczeniem aktywnym u pacjentów z pogorszeniem widzenia spowodowanym cukrzycowym obrzękiem plamki żółtej.Łącznie włączono do tych badań 496 pacjentów (336 aktywnych i 160 osób z grupy kontrolnej), większość miała cukrzycę typu II, 28 leczonych pacjentów miało cukrzycę typu I.
W fazie II badania D2201 (RESOLVE) 151 pacjentów leczono ranibizumabem (6 mg/ml, n = 51, 10 mg/ml, n = 51) lub pozorowanym (n = 49) jednym „wstrzyknięciem do ciała szklistego na miesiąc”. do czasu spełnienia wcześniej zdefiniowanych kryteriów Dawkę początkową ranibizumabu (0,3 mg lub 0,5 mg) można było podwoić w dowolnym momencie badania po pierwszym wstrzyknięciu. Fotokoagulacja laserowa była dozwolona jako leczenie ratunkowe od 3. miesiąca w obu ramionach leczenia. składał się z dwóch części: eksploracyjnej (pierwszych 42 pacjentów odwiedzonych w 6. miesiącu) oraz potwierdzającej (pozostałych 109 pacjentów odwiedzonych w 12. miesiącu).
Kluczowe wnioski z części potwierdzającej badania (2/3 pacjentów) podsumowano w Tabeli 3.
Tabela 3 Wyniki w 12. miesiącu w badaniu D2201 (RESOLVE) (całkowita populacja badania)
ap
W badaniu III fazy D2301 (RESTORE) 345 pacjentów z zaburzeniami widzenia spowodowanymi obrzękiem plamki zostało losowo przydzielonych do otrzymywania „wstrzyknięcia do ciała szklistego 0,5 mg ranibizumabu w monoterapii i pozorowanej laserowej fotokoagulacji (n = 116) lub kombinacji 0,5 mg ranibizumabu oraz fotokoagulacja laserowa (n=118) lub pozorowane wstrzyknięcie i fotokoagulacja laserowa (n=111). Leczenie ranibizumabem rozpoczęto od comiesięcznych wstrzyknięć do ciała szklistego i kontynuowano aż do ustabilizowania się ostrości wzroku przez co najmniej trzy kolejne comiesięczne kontrole Leczenie wznowiono, gdy zaobserwowano zmniejszenie BCVA z powodu progresji DME. co najmniej 30 minut przed wstrzyknięciem ranibizumabu, a następnie w razie potrzeby na podstawie kryteriów ETDRS.
Kluczowe ustalenia podsumowano w tabeli 4 i na rysunku 2.
Tabela 4 Wyniki w 12. miesiącu w badaniu D2301 (RESTORE)
ap
Efekt był spójny w większości podgrup, jednak osoby z dość wysoką wyjściową wartością BCVA (> 73 litery) z obrzękiem plamki i centralną grubością siatkówki
Poprawie ostrości wzroku w miesiącu 12. obserwowanej po podaniu produktu Lucentis 0,5 mg towarzyszyły zgłaszane przez pacjentów korzyści z głównych funkcji związanych z widzeniem, mierzone za pomocą kwestionariusza National Eye Institute Visual Function Questionnaire (VFQ-25). ustalone w podklasach tego kwestionariusza.Różnicę między produktem Lucentis 0,5 mg a grupą kontrolną oceniano przy wartości p 0,0137 (ranibizumab mono) i 0,0041 (ranibizumab + laser) dla złożonej punktacji VFQ-25.
W obu badaniach poprawie widzenia towarzyszyło ciągłe zmniejszanie się obrzęku plamki mierzonego jako centralna grubość siatkówki (CRT).
Leczenie zaburzeń widzenia spowodowanych obrzękiem plamki wtórnym do RVO
Bezpieczeństwo kliniczne i skuteczność produktu Lucentis u pacjentów z zaburzeniami widzenia spowodowanymi obrzękiem plamki wtórnym do RVO oceniano w randomizowanych, podwójnie zaślepionych, kontrolowanych badaniach: BRAVO i CRUISE, do których rekrutowano pacjentów z BRVO (n = 397) i CRVO (n = 392). W obu badaniach pacjenci otrzymywali 0,3 mg lub 0,5 mg ranibizumabu w postaci wstrzyknięć do ciała szklistego lub pozorowanych. Po 6 miesiącach pacjenci z grupy kontrolnej pozorowanej zostali przeniesieni do grupy ranibizumabu w dawce 0,5 mg. W badaniu BRAVO fotokoagulacja laserowa jako leczenie ratunkowe był dozwolony we wszystkich ramionach od 3. miesiąca.
Kluczowe wyniki badań BRAVO i CRUISE przedstawiono w tabelach 5 i 6
Tabela 5 Wyniki w 6. i 12. miesiącu (BRAVO)
ap
Tabela 6 Wyniki w 6. i 12. miesiącu (REJS)
ap
W obu badaniach poprawie wzroku towarzyszyło ciągłe i znaczne zmniejszenie obrzęku plamki mierzonego grubością siatkówki centralnej.
U pacjentów z BRVO (badanie BRAVO i rozszerzenie badania HORIZON): Po 2 latach pacjenci, którzy byli leczeni wstrzyknięciami pozorowanymi w ciągu pierwszych 6 miesięcy, a następnie przeszli na leczenie ranibizumabem, uzyskali przyrost AV (& symp; 15 liter) porównywalny do tego pacjentów, którzy byli leczeni ranibizumabem od rozpoczęcia badania (& symp; 16 liter).Jednak liczba pacjentów, którzy ukończyli 2 lata, była ograniczona i w badaniu HORIZON zaplanowano tylko wizyty kwartalne. Istnieją wystarczające dowody, aby zakończyć z zaleceniami dotyczącymi kiedy należy rozpocząć leczenie ranibizumabem u pacjentów z BRVO.
U pacjentów z CRVO (badanie CRUISE i rozszerzenie badania HORIZON): Po 2 latach pacjenci, którzy w ciągu pierwszych 6 miesięcy byli leczeni wstrzyknięciami pozorowanymi, a następnie przeszli na leczenie ranibizumabem, nie wykazali wzrostu AV (& symp; 6 liter) w porównaniu z pacjentów, którzy byli leczeni ranibizumabem od rozpoczęcia badania (& symp; 12 liter).
Poprawie ostrości wzroku obserwowanej podczas leczenia ranibizumabem w miesiącach 6 i 12 towarzyszyły korzyści zgłaszane przez pacjentów, mierzone za pomocą podgrupy kwestionariusza funkcji wzrokowych National Eye Institute (NEI VFQ-25) dla czynności bliskich i dalekich. Różnica między produktem Lucentis 0,5 mg a grupa kontrolna znajdowała się między wartościami p od 0,02 do 0,0002.
Leczenie zaburzeń widzenia spowodowanych CNV wtórną do PM
Bezpieczeństwo i skuteczność kliniczną produktu Lucentis u pacjentów z zaburzeniami widzenia spowodowanymi CNV w PM zostały zweryfikowane na podstawie danych z 12 miesięcy z kluczowego, randomizowanego, podwójnie zaślepionego, kontrolowanego badania F2301 (RADIANCE).To badanie miało na celu ocenę dwóch różnych schematów dawkowania ranibizumabu w dawce 0,5 mg podawanego we wstrzyknięciu do ciała szklistego w porównaniu z PDT z werteporfiną (vPDT, terapia fotodynamiczna Visudyne).277 pacjentów przydzielono losowo do jednego z następujących ramion:
• Grupa I (ranibizumab 0,5 mg, schemat leczenia określony kryteriami „stabilności” zdefiniowanymi jako brak zmiany BCVA w porównaniu z ocenami z poprzednich dwóch miesięcy).
• Grupa II (ranibizumab 0,5 mg, schemat leczenia określony kryteriami „aktywności choroby” zdefiniowanymi jako zaburzenia widzenia przypisywane płynowi wewnątrz- lub podsiatkówkowemu lub aktywnym przeciekom spowodowanym zmianami CNV potwierdzonymi przez OCT i/lub AF) .
• Grupa III (pacjenci leczeni vPDT – z możliwością leczenia ranibizumabem od 3 miesiąca).
W ciągu 12 miesięcy badania pacjenci otrzymali średnio 4,6 iniekcji (zakres 1-11) w grupie I i 3,5 iniekcji (zakres 1-12) w grupie II. Wśród pacjentów należących do grupy II, co odzwierciedla zalecane dawkowanie (patrz punkt 4.2), 50,9% pacjentów przeszło leczenie 1 do 2 wstrzyknięć, 34,5% 3 do 5 wstrzyknięć, a 14,7% wykonało 6 do 12 wstrzyknięć w ciągu 12 miesięcy badania . 62,9% pacjentów z grupy II nie wymagało wstrzyknięć w ciągu drugich 6 miesięcy badania.
Kluczowe wyniki badania RADIANCE podsumowano w Tabeli 7 i na Ryc. 5.
Tabela 7 Wyniki w 3. i 12. miesiącu (RADIANCE)
ap
b Kontrola porównawcza do 3. miesiąca Pacjenci przydzieleni losowo do grupy otrzymującej vPDT kwalifikowali się do leczenia ranibizumabem w 3. miesiącu (w grupie III 38 pacjentów otrzymywało ranibizumab w 3. miesiącu)
Poprawie widzenia towarzyszyło zmniejszenie grubości środkowej siatkówki.
W porównaniu z grupą leczoną vPDT pacjenci w grupach leczonych ranibizumabem zgłaszali korzyści (wartość p
Populacja pediatryczna
Bezpieczeństwo i skuteczność ranibizumabu u dzieci nie zostały jeszcze ustalone.
Europejska Agencja Leków uchyliła obowiązek przedstawienia wyników badań produktu Lucentis we wszystkich podgrupach populacji pediatrycznej z wysiękową postacią AMD, zaburzeniami widzenia spowodowanymi DME, zaburzeniami widzenia spowodowanymi wtórnym obrzękiem plamki spowodowanym RVO oraz zaburzeniami widzenia spowodowanymi CNV wtórną do RVO. PM (informacje dotyczące stosowania u dzieci, patrz punkt 4.2).
05.2 Właściwości farmakokinetyczne
Po comiesięcznym podawaniu produktu Lucentis do ciała szklistego pacjentom z wysiękową postacią AMD stężenia ranibizumabu w surowicy były na ogół niskie, przy czym stężenia maksymalne (Cmax) były zazwyczaj poniżej stężenia ranibizumabu wymaganego do zahamowania aktywności biologicznej VEGF o 50% (11-27 ng/ml, oceniane w teście in vitro proliferacja komórek). Cmax było proporcjonalne do dawki w całym zakresie dawek od 0,05 do 1,0 mg/oko U ograniczonej liczby pacjentów z DME wykryte stężenia w surowicy wskazują, że nie można wykluczyć nieco większej ekspozycji ogólnoustrojowej niż obserwowana u pacjentów z wysiękową postacią AMD. Stężenia ranibizumabu w surowicy pacjentów z RVO były podobne lub nieznacznie wyższe niż te obserwowane u pacjentów z wysiękową postacią AMD.
Na podstawie analizy farmakokinetyki populacyjnej i klirensu ranibizumabu z surowicy u pacjentów z wysiękową postacią AMD leczonych dawką 0,5 mg, średni okres półtrwania ranibizumabu w fazie eliminacji do ciała szklistego wynosi około 9 dni. Oczekuje się, że w czasie comiesięcznego podawania do ciała szklistego produktu Lucentis 0,5 mg/oko stężenie Cmin ranibizumabu w surowicy, osiągane około 1 dzień po podaniu dawki, będzie na ogół wynosić od 0,79 do 2,90 ng/ml, podczas gdy oczekuje się, że Cmin na ogół waha się między 0,07 i 0,49 ng / ml. Szacuje się, że stężenia ranibizumabu w surowicy są około 90 000 razy niższe niż stężenia w ciele szklistym.
Pacjenci z niewydolnością nerek: Nie przeprowadzono konwencjonalnych badań oceniających farmakokinetykę produktu Lucentis u pacjentów z niewydolnością nerek. W „analizie farmakokinetycznej w populacji pacjentów z wysiękową postacią AMD, 68% (136 z 200) pacjentów miało” niewydolność nerek (46,5% łagodna [50-80 ml/min], 20% umiarkowana [30-50 ml/min] min] i 15% ciężki [klirens ogólnoustrojowy był nieco niższy, ale nie było to istotne klinicznie.
Pacjenci z niewydolnością wątroby: Nie przeprowadzono konwencjonalnych badań oceniających farmakokinetykę produktu Lucentis u pacjentów z niewydolnością wątroby.
05.3 Przedkliniczne dane o bezpieczeństwie
Obustronne podawanie do ciała szklistego ranibizumabu małpom cynomolgus w dawkach od 0,25 mg/oko do 2,0 mg/oko raz na 2 tygodnie przez okres do 26 tygodni powodowało zależne od dawki działanie oczne.
Wewnątrzgałkowe, zależne od dawki nasilenie zaostrzenia i komórek wystąpiło w komorze przedniej, osiągając maksimum 2 dni po wstrzyknięciu.Nasilenie odpowiedzi zapalnej zwykle zmniejsza się po kolejnych wstrzyknięciach lub w okresie zdrowienia.W odcinku tylnym wystąpiły nacieki komórkowe i męty w ciele szklistym, które również wykazywało tendencję do zależności od dawki i ogólnie utrzymywało się do końca okresu leczenia.W badaniu trwającym 26 tygodni nasilenie zapalenia ciała szklistego zwiększało się wraz z liczbą wstrzyknięć. Jednak po okresie rekonwalescencji zaobserwowano odwracalność. Charakter i czas trwania zapalenia tylnego odcinka oka wskazują na odpowiedź przeciwciał o podłożu immunologicznym, która może być klinicznie nieistotna.U niektórych zwierząt zaobserwowano tworzenie się zaćmy po stosunkowo długim okresie intensywnego zapalenia, co sugeruje, że zmiany soczewki były wtórne. do ciężkiego stanu zapalnego. Po podaniu, niezależnie od dawki, po wstrzyknięciach do ciała szklistego obserwowano przemijający wzrost ciśnienia wewnątrzgałkowego.
Mikroskopowe zmiany oczne były związane ze stanem zapalnym i nie wskazywały na procesy zwyrodnieniowe, w tarczy nerwu wzrokowego niektórych oczu obserwowano zmiany zapalne ziarniniakowe, które w okresie rekonwalescencji zmniejszyły się, a w niektórych przypadkach ustąpiły.
Nie stwierdzono objawów toksyczności ogólnoustrojowej po podaniu do ciała szklistego. W podgrupie leczonych zwierząt wykryto przeciwciała w surowicy i ciele szklistym przeciwko ranibizumabowi.
Brak danych dotyczących rakotwórczości lub mutagenności.
U ciężarnych małp wstrzyknięcie do ciała szklistego ranibizumabu skutkujące maksymalną ekspozycją ogólnoustrojową 0,9-7 razy najgorszą ekspozycją kliniczną nie powodowało toksyczności rozwojowej ani teratogenności i nie miało wpływu na masę ciała ani strukturę łożyska, chociaż ranibizumab należy traktować jako potencjalnie teratogenny i embriotoksyczny/fetotoksyczny w oparciu o działanie farmakologiczne.
Brak pośredniczącego wpływu ranibizumabu na rozwój zarodka/płodu jest prawdopodobnie związany głównie z niezdolnością fragmentu Fab do przenikania przez łożysko. Opisano jednak przypadek z wysokimi stężeniami ranibizumabu w surowicy matki i obecnością ranibizumabu w surowicy płodu, co sugeruje, że przeciwciało przeciw ranibizumabowi działało jak białko (zawierające region FC), które transportuje ranibizumab, zmniejszając w ten sposób jego eliminację z surowicy matki i umożliwienie jej przeniesienia do łożyska. Ponieważ badania dotyczące rozwoju zarodka/płodu przeprowadzono na zdrowych ciężarnych zwierzętach, a niektóre choroby (takie jak cukrzyca) mogą modyfikować przepuszczalność fragmentu Fab przez łożysko, badanie należy interpretować z ostrożnością.
06.0 INFORMACJE FARMACEUTYCZNE
06.1 Zaróbki
dihydrat α, α-trehalozy
Monohydrat chlorowodorku histydyny
Histydyna
Polisorbat 20
Woda do wstrzykiwań
06.2 Niekompatybilność
Ze względu na brak badań zgodności, tego produktu leczniczego nie wolno mieszać z innymi produktami leczniczymi.
06.3 Okres ważności
3 lata
06.4 Specjalne środki ostrożności przy przechowywaniu
Przechowywać w lodówce (2°C - 8°C).
Nie zamrażać.
Fiolkę przechowywać w opakowaniu zewnętrznym w celu ochrony leku przed światłem.
06.5 Rodzaj opakowania bezpośredniego i zawartość opakowania
0,23 ml jałowego roztworu w fiolce (szkło typu I) z korkiem (guma chlorobutylowa), 1 tępa igła z filtrem (18G x 1½", 1,2 mm x 40 mm, 5 mcm), 1 igła iniekcyjna (30G x ½", 0,3 mm x 13 mm) i 1 strzykawkę (polipropylen) (1 ml). Opakowanie zawiera 1 fiolkę.
06.6 Instrukcje użytkowania i obsługi
Fiolka, igła iniekcyjna, igła z filtrem i strzykawka są przeznaczone wyłącznie do jednorazowego użytku. Ponowne użycie może spowodować infekcję lub inną chorobę / uraz. Wszystkie składniki są sterylne. Nie wolno używać żadnego elementu, którego opakowanie nosi ślady uszkodzenia lub manipulacji. Nie można zagwarantować sterylności, jeśli uszczelnienie opakowania komponentu nie jest nienaruszone.
Aby przygotować lek Lucentis do wstrzyknięcia do ciała szklistego, należy postępować zgodnie z poniższymi instrukcjami:
1. Zdezynfekować zewnętrzną część gumowego korka fiolki przed pobraniem.
2. Aseptycznie przymocować igłę z filtrem 5 mcm (18G x 1½", 1,2 mm x 40 mm, dołączona) do 1 ml strzykawki (w zestawie). Wsunąć tępą igłę z filtrem w środek nasadki, aż dotknie dna fiolki.
3. Pobrać cały płyn z fiolki, trzymając ją w pozycji pionowej, lekko przechyloną, aby ułatwić całkowite pobranie.
4. Upewnić się, że tłok strzykawki jest odciągnięty wystarczająco daleko podczas opróżniania fiolki, aby całkowicie opróżnić igłę z filtrem.
5. Pozostawić tępą igłę z filtrem w fiolce i wyjąć z niej strzykawkę.Wyrzucić igłę z filtrem po pobraniu zawartości fiolki i nie używać jej do wstrzyknięcia do ciała szklistego.
6. Przymocować igłę iniekcyjną (30G x ½”, 0,3mm x 13mm, w zestawie) bezpiecznie i aseptycznie do strzykawki.
7. Ostrożnie zdjąć nasadkę z igły iniekcyjnej bez odłączania igły iniekcyjnej od strzykawki.
Uwaga: Przytrzymaj żółtą podstawę igły do wstrzykiwań podczas zdejmowania nasadki.
8. Ostrożnie usunąć powietrze ze strzykawki i dostosować dawkę do 0,05 ml oznaczonego na strzykawce.Strzykawka jest gotowa do wstrzyknięcia.
Uwaga: Nie czyścić igły do wstrzykiwań i nie odciągać tłoka.
Po wstrzyknięciu nie zakrywać igły ani nie odłączać jej od strzykawki. Zużytą strzykawkę wraz z igłą wyrzucić do odpowiedniego pojemnika lub zgodnie z lokalnymi przepisami.
07.0 PODMIOT POZWOLENIA NA DOPUSZCZENIE DO OBROTU
Novartis Europharm Limited
Droga Wimblehurst
Horszam
West Sussex, RH12 5AB
Wielka Brytania
08.0 NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
UE / 1/06/374/001
037608027
09.0 DATA PIERWSZEGO ZEZWOLENIA LUB PRZEDŁUŻENIA ZEZWOLENIA
Data pierwszego zezwolenia: 22 stycznia 2007 r.
Data ostatniego przedłużenia: 24 stycznia 2012 r.
10.0 DATA ZMIAN TEKSTU
05/2014