Ogólność
DNA mitochondrialne, inaczej mtDNA, to kwas dezoksyrybonukleinowy, który znajduje się wewnątrz mitochondriów, czyli organelli komórek eukariotycznych, odpowiedzialnych za bardzo ważny komórkowy proces fosforylacji oksydacyjnej.

Ma jednak również pewne osobliwości, zarówno konstrukcyjne, jak i funkcjonalne, które czynią go wyjątkowym w swoim rodzaju. Do tych osobliwości należą: kołowość podwójnej nici nukleotydów, zawartość genów (czyli tylko 37 elementów) i prawie całkowity brak niekodujących sekwencji nukleotydowych.
DNA mitochondrialne pełni fundamentalną funkcję dla przetrwania komórek: wytwarza enzymy niezbędne do realizacji fosforylacji oksydacyjnej.
Czym jest mitochondrialne DNA?
DNA mitochondrialne lub mtDNA to DNA znajdujące się w mitochondriach.
Mitochondria to duże organelle komórkowe, typowe dla organizmów eukariotycznych, które przekształcają energię chemiczną zawartą w pożywieniu w ATP, który jest formą energii, która może być wykorzystywana przez komórki.
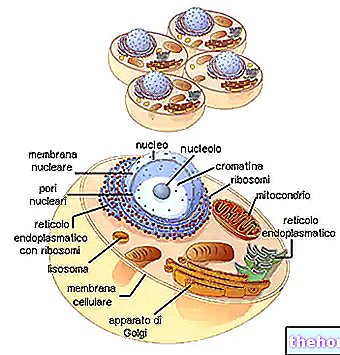
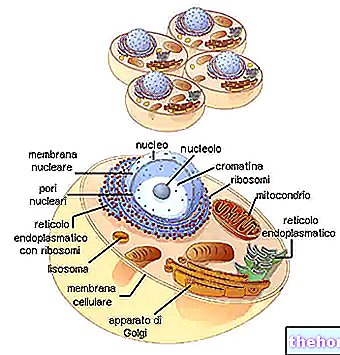
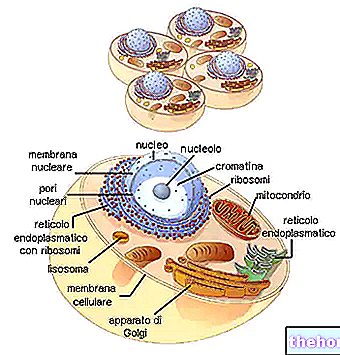
TŁO STRUKTURY I FUNKCJONOWANIA MITOCHONDRONÓW
W cytoplazmie znajdują się mitochondria o kształcie cylindrycznym, nitkowatym lub ziarnistym, zajmując prawie 25% objętości tej ostatniej.
Mają dwie dwuwarstwowe błony fosfolipidowe, jedną bardziej zewnętrzną i jedną bardziej wewnętrzną.
Najbardziej zewnętrzna błona, znana jako zewnętrzna błona mitochondrialna, reprezentuje obwód każdego mitochondrium i zawiera białka transportowe (poryny i inne), dzięki czemu jest przepuszczalna dla cząsteczek o rozmiarze równym lub mniejszym niż 5000 daltonów.
Najbardziej wewnętrzna błona, znana jako wewnętrzna błona mitochondrialna, zawiera wszystkie składniki enzymatyczne (lub enzymatyczne) i koenzymu, niezbędne do syntezy ATP i definiuje przestrzeń centralną, zwaną macierzą.
W przeciwieństwie do błony zewnętrznej, wewnętrzna błona mitochondrialna ma liczne wgłębienia – tak zwane grzbiety – które zwiększają jej całkowitą powierzchnię.
Pomiędzy dwiema błonami mitochondrialnymi znajduje się przestrzeń prawie 60-80 angstremów (A), którą nazywamy przestrzenią międzybłonową, która ma budowę bardzo zbliżoną do cytoplazmy.
Synteza ATP prowadzona przez mitochondria to bardzo złożony proces, który biolodzy utożsamiają z terminem fosforylacja oksydacyjna.
PRECYZYJNA LOKALIZACJA MITOCHONDRALNEGO DNA I ILOŚCI

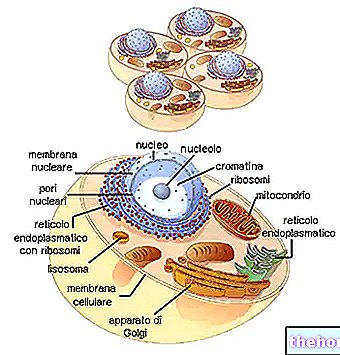
Rysunek: ludzkie mitochondrium.
DNA mitochondrialne znajduje się w macierzy mitochondrialnej, czyli w przestrzeni ograniczonej wewnętrzną błoną mitochondrialną.
Według wiarygodnych badań naukowych każde mitochondrium może zawierać od 2 do 12 kopii mitochondrialnego DNA.
Biorąc pod uwagę fakt, że w ludzkim ciele niektóre komórki mogą zawierać w sobie kilka tysięcy mitochondriów, łączna liczba kopii mitochondrialnego DNA w pojedynczej komórce człowieka może sięgać nawet 20 000 jednostek.
Proszę zanotować: liczba mitochondriów w komórkach ludzkich różni się w zależności od typu komórki. Na przykład hepatocyty (tj. komórki wątroby) mogą zawierać od 1000 do 2000 mitochondriów każdy, podczas gdy erytrocyty (tj. czerwone krwinki) są ich całkowicie pozbawione.
Struktura
Ogólna struktura cząsteczki mitochondrialnego DNA przypomina ogólną strukturę jądrowego DNA, czyli dziedzictwo genetyczne obecne w jądrze komórek eukariotycznych.
W rzeczy samej, analogicznie do jądrowego DNA:
- DNA mitochondrialne to biopolimer składający się z dwóch długich nici nukleotydów. Nukleotydy to cząsteczki organiczne powstałe z połączenia trzech elementów: cukru o 5 atomach węgla (w przypadku DNA dezoksyryboza), zasady azotowej i grupy fosforanowej.
- Każdy nukleotyd mitochondrialnego DNA wiąże się z następnym nukleotydem tej samej nici za pomocą wiązania fosfodiestrowego między węglem 3 jego dezoksyrybozy i grupą fosforanową następnego nukleotydu.
- Dwie nici mitochondrialnego DNA mają przeciwne orientacje, przy czym koniec jednej wchodzi w interakcję z końcem drugiej i odwrotnie.Ten szczególny układ jest znany jako układ antyrównoległy (lub orientacja antyrównoległa).
- Dwie nici mitochondrialnego DNA oddziałują ze sobą poprzez zasady azotowe.
W szczególności każda zasada azotowa każdego włókna tworzy wiązania wodorowe z jedną i tylko jedną zasadą azotową obecną na drugim włóknie.
Ten rodzaj interakcji nazywa się „parowaniem zasad azotowych” lub „parą zasad azotowych”. - Azotowe zasady mitochondrialnego DNA to adenina, tymina, cytozyna i guanina.
Pary, do których prowadzą te zasady azotowe, nie są przypadkowe, ale bardzo specyficzne: adenina oddziałuje tylko z tyminą, podczas gdy cytozyna oddziałuje tylko z guaniną. - DNA mitochondrialne jest domem dla genów (lub sekwencji genów). Geny to sekwencje mniej lub bardziej długich nukleotydów o dobrze zdefiniowanym znaczeniu biologicznym. W większości przypadków powodują powstawanie białek.

STRUKTURALNE SZCZEGÓLNOŚCI MITOCHONDRALNEGO DNA
Poza wyżej wymienionymi analogiami, ludzki mitochondrialny DNA ma pewne cechy strukturalne, które znacznie odróżniają go od ludzkiego DNA jądrowego.
Po pierwsze, jest to cząsteczka kołowa, podczas gdy jądrowe DNA jest cząsteczką liniową.
Tak więc ma 16 569 par zasad azotowych, podczas gdy jądrowy DNA ma aż 3,3 miliarda.
Zawiera 37 genów, podczas gdy jądrowy DNA wydaje się zawierać od 20 000 do 25 000.
Nie jest zorganizowany w chromosomy, podczas gdy jądrowy DNA jest podzielony na 23 chromosomy i tworzy, z pewnymi specyficznymi białkami, substancję zwaną chromatyną.
Wreszcie zawiera serię nukleotydów, które uczestniczą w dwóch genach jednocześnie, podczas gdy jądrowy DNA ma geny, których sekwencje nukleotydów są dobrze zdefiniowane i różnią się od siebie.
Początek
Mitochondrialny DNA najprawdopodobniej ma pochodzenie „bakteryjne”.
W rzeczywistości, na podstawie licznych niezależnych badań, biolodzy molekularni uważają, że komórkowa obecność mitochondrialnego DNA jest wynikiem wbudowania przez przodków komórek eukariotycznych niezależnych organizmów bakteryjnych, bardzo podobnych do mitochondriów.
To ciekawe odkrycie tylko częściowo zadziwiło społeczność naukową, ponieważ DNA obecne w bakteriach jest na ogół kołową nicią nukleotydową, taką jak DNA mitochondrialne.
Teoria, zgodnie z którą mitochondria i mitochondrialne DNA mają „bakteryjne pochodzenie”, wzięła nazwę „teoria endosymbiozy” od słowa „endosymbioza”. Krótko mówiąc, w biologii termin „endosymbioza” wskazuje na współpracę między dwoma organizmami, która obejmuje „włączenie jednego w drugie w celu uzyskania pewnej przewagi.
Ciekawość
Według wiarygodnych badań naukowych, w toku ewolucji wiele genów bakteryjnych obecnych w przyszłym mitochondrialnym DNA zmieniłoby lokalizację, przenosząc się do DNA jądrowego.
Innymi słowy, na początku endosymbiozy niektóre geny obecne w DNA jądrowym znajdowały się w DNA tych organizmów bakteryjnych, które później stały się mitochondriami.
Aby poprzeć teorię dotyczącą przesunięcia genów między mitochondrialnym DNA a jądrowym DNA, jest obserwacja, że niektóre geny wywodzą się z mitochondrialnego DNA u niektórych gatunków, a jądrowego DNA u innych.
Funkcjonować
DNA mitochondrialne wytwarza enzymy (tj. białka), niezbędne do prawidłowej realizacji delikatnego procesu fosforylacji oksydacyjnej.
Instrukcje dotyczące syntezy tych enzymów znajdują się w 37 genach tworzących genom mitochondrialnego DNA.
CO KODUJĄ MITOCHONDRALNE GENY DNA: SZCZEGÓŁY
37 genów mitochondrialnego DNA koduje: białka, tRNA i rRNA.
W szczególności:
- 13 koduje 13 białek odpowiedzialnych za przeprowadzenie fosforylacji oksydacyjnej
- Kod 22 dla 22 cząsteczek tRNA
- 2 kodują 2 cząsteczki rRNA
Cząsteczki tRNA i rRNA mają fundamentalne znaczenie dla syntezy wyżej wymienionych 13 białek, ponieważ tworzą maszynerię regulującą ich produkcję.
Innymi słowy, mitochondrialne DNA posiada informacje potrzebne do wytworzenia pewnego zestawu białek i narzędzi niezbędnych do ich syntezy.
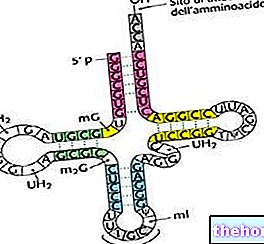
Czym są RNA, tRNA i rRNA?
RNA lub kwas rybonukleinowy to kwas nukleinowy, który odgrywa fundamentalną rolę w tworzeniu białek, począwszy od DNA.
Ogólnie rzecz biorąc, jednoniciowa sieć SSN może występować w różnych formach (lub typach), w zależności od konkretnej funkcji, do której jest delegowana.
TRNA i rRNA to dwie z tych możliwych form.
tRNA służy do dodawania aminokwasów podczas procesu tworzenia białek.Aminokwasy są jednostkami molekularnymi, które tworzą białka.
rRNA tworzy rybosomy, czyli struktury komórkowe, w których zachodzi synteza białek.
Aby poznać szczegółowo sieć SSN i jej funkcje, czytelnicy mogą kliknąć tutaj.
SZCZEGÓŁY FUNKCJONALNE MITOCHONDRALNEGO DNA
Z funkcjonalnego punktu widzenia mitochondrialne DNA ma pewne szczególne cechy, które wyraźnie odróżniają go od DNA jądrowego.
Oto, z czego składają się te szczególne cechy:
- DNA mitochondrialne jest częściowo niezależne, w tym sensie, że wymaga interwencji niektórych białek syntetyzowanych z DNA jądrowego.
Z drugiej strony jądrowe DNA jest całkowicie autonomiczne i samodzielnie wytwarza wszystko, czego potrzebuje do prawidłowego wykonywania swoich zadań. - DNA mitochondrialne ma nieco inny kod genetyczny niż DNA jądrowy.Prowadzi to do szeregu różnic w tworzeniu białek: jeśli pewna sekwencja nukleotydów w jądrowym DNA prowadzi do powstania określonego białka, to ta sama sekwencja w mitochondrialnym DNA prowadzi do powstania nieco innego białka.
- DNA mitochondrialne ma bardzo niewiele niekodujących sekwencji nukleotydowych, to znaczy nie wytwarza żadnych białek, tRNA ani rRNA. W ujęciu procentowym tylko 3% mitochondrialnego DNA nie koduje.
Z drugiej strony jądrowe DNA koduje tylko 7%, a więc zawiera wiele niekodujących sekwencji nukleotydowych (aż 93%).
Tabela: podsumowanie różnic między ludzkim mitochondrialnym DNA a ludzkim jądrowym DNA.
Mitochondrialny DNA
Jądrowe DNA
- Jest okrągły
- Jest liniowy
- Ma w sumie 16 569 par zasad azotowych
- Ma łącznie 3,3 miliarda par zasad azotowych
- Zawiera łącznie 37 genów
- Zawiera od 20 000 do 25 000 genów
- Do prawidłowego funkcjonowania potrzebuje wsparcia niektórych produktów genów, pochodzących z jądrowego DNA
- Jest autonomiczny i sam wytwarza wszystko, czego potrzebuje do prawidłowego wykonywania swoich funkcji
- Może występować w kilku kopiach w każdym pojedynczym mitochondrium
- Jest unikalny, to znaczy występuje tylko w jednym egzemplarzu i znajduje się w rdzeniu
- 97% składającej się na nią sekwencji nukleotydowej koduje
- Tylko 7% składającej się na nią sekwencji nukleotydowej koduje
- Nie jest zorganizowany w chromosomy
- Jest podzielony na 23 chromosomy
- Wykorzystuje kod genetyczny nieco inny niż, że tak powiem, „tradycyjny”
- Użyj „tradycyjnego” kodu genetycznego
- Jego dziedzictwo jest matczyne
- Jego dziedzictwo jest w połowie matczyne i w połowie ojcowskie
- Niektóre z jego nukleotydów uczestniczą jednocześnie w dwóch genach
- Sekwencje nukleotydów tworzących geny są dobrze od siebie odróżnione
Dziedzictwo
Dziedziczenie mitochondrialnego DNA jest ściśle matczyne.
Oznacza to, że w przypadku pary rodziców to kobieta przekazuje mitochondrialne DNA potomstwu (tj. dzieciom).
W sposób całkowicie odwrotny do powyższego, dziedziczenie jądrowego DNA jest w połowie matczyne i w połowie ojcowskie, innymi słowy, oboje rodzice w równym stopniu przyczyniają się do przenoszenia DNA jądrowego u potomstwa.
Proszę zanotować: matczyne dziedziczenie mitochondrialnego DNA obejmuje również strukturę mitochondrialną. Stąd mitochondria obecne u osobnika są matczyne.
Powiązane patologie
Przesłanka: Mutacja genetyczna to trwała zmiana w sekwencji nukleotydów, które tworzą jądrowy lub mitochondrialny gen DNA.
Zazwyczaj obecność mutacji genetycznej powoduje „zmianę lub utratę normalnej funkcji zaangażowanego genu.
Obecność mutacji w genach mitochondrialnego DNA może prowadzić do wielu chorób, w tym:
- Dziedziczna neuropatia wzrokowa Lebera
- Zespół Kearnsa-Sayre'a
- Zespół Leigh
- Niedobór oksydazy cytochromu C
- Postępująca oftalmoplegia zewnętrzna
- Zespół Pearsona
- Encefalomiopatia mitochondrialna z kwasicą mleczanową i epizodami udaropodobnymi (zespół MELAS)
- Cukrzyca z głuchotą przenoszoną przez matkę
- Padaczka miokloniczna z nieregularnymi czerwonymi włóknami
Jeśli chodzi o stany patologiczne związane z jedną lub więcej mutacjami mitochondrialnego DNA, należy wyjaśnić dwa aspekty.
Po pierwsze, ciężkość choroby zależy od ilościowego związku między zmutowanym mitochondrialnym DNA a zdrowym, normalnym mitochondrialnym DNA. Jeśli liczba zmutowanych mitochondrialnych DNA jest znacznie większa niż zdrowych DNA, wynikowy stan będzie bardziej dotkliwy.
Po drugie, mutacje w mitochondrialnym DNA dotyczą tylko niektórych tkanek organizmu, w szczególności tych, które wymagają dużych ilości ATP powstającego w procesie fosforylacji oksydacyjnej, co jest całkiem zrozumiałe: na więcej niż jedną wadę mitochondrialnego DNA najbardziej potrzebują komórki funkcję, jaką normalnie spełnia mitochondrialne DNA.
DZIEDZICZNA NEUROPATIA OPTYCZNA LEBERA
Dziedziczna neuropatia nerwu wzrokowego Lebera powstaje w wyniku mutacji aż czterech genów mitochondrialnego DNA. Geny te zawierają informację, która prowadzi do syntezy tzw. kompleksu I (lub reduktazy tlenku NADH), jednego z różnych enzymów biorących udział w procesie fosforylacji oksydacyjnej.
Objawy patologii polegają na postępującej degeneracji nerwu wzrokowego i stopniowej utracie wzroku.
ZESPÓŁ KEARNSA-SAYREA
Zespół Kearnsa-Sayre'a pojawia się z powodu braku dużej ilości mitochondrialnego DNA (Uwaga: brak określonej sekwencji nukleotydowej nazywa się delecją).
U osób z zespołem Kearnsa-Sayre'a rozwija się oftalmoplegia (całkowity lub częściowy paraliż mięśni okoruchowych), postać retinopatii i zaburzenia rytmu serca (blok przedsionkowo-komorowy).
ZESPÓŁ LEIGHA
Zespół Leigh powstaje w wyniku mutacji mitochondrialnego DNA, które mogą wpływać na białko syntazy ATP (zwane również kompleksem V) i / lub niektóre tRNA.
Zespół Leigh jest postępującą chorobą neurologiczną, która pojawia się w okresie niemowlęcym lub dziecięcym i jest odpowiedzialna za: opóźnienie rozwoju, osłabienie mięśni, neuropatię obwodową, zaburzenia motoryczne, trudności w oddychaniu i oftalmoplegię.
NIEDOBOR CYTOCHROMU C OKSYDAZY
Niedobór oksydazy cytochromu C występuje z powodu mutacji co najmniej 3 genów mitochondrialnego DNA. Geny te są niezbędne do prawidłowej syntezy enzymu oksydazy cytochromu C (lub kompleksu IV), biorącego udział w procesie fosforylacji oksydacyjnej.
Typowe objawy niedoboru oksydazy cytochromu C to: dysfunkcja mięśni szkieletowych, dysfunkcja serca, dysfunkcja nerek i dysfunkcja wątroby.
POSTĘPUJĄCA ZEWNĘTRZNA OFTALMOPLEGIA
Postępująca oftalmoplegia zewnętrzna wynika z braku znacznej liczby nukleotydów mitochondrialnego DNA (delecja)
Przy progresywnym charakterze (jak można się domyślić z nazwy), patologia ta powoduje paraliż mięśni okulomotorycznych, w konsekwencji opadanie powiek i znaczne problemy ze wzrokiem.
ZESPÓŁ PEARSONA
Zespół Pearsona pojawia się po wyraźnej delecji mitochondrialnego DNA, podobnie jak postępująca oftalmoplegia zewnętrzna i zespół Kearnsa-Sayre'a.
Typowe objawy zespołu Pearsona to: niedokrwistość syderoblastyczna, dysfunkcja trzustki (np. cukrzyca insulinozależna), ubytki neurologiczne i zaburzenia mięśniowe.
Zespół Pearsona zwykle powoduje, że osoba dotknięta chorobą umiera w młodym wieku. W rzeczywistości osoby dotknięte tą patologią rzadko osiągają dorosłość.
ZESPÓŁ MELAS
Zespół MELAS, znany również jako encefalomiopatia mitochondrialna z kwasicą mleczanową i epizodami udaropodobnymi, powstaje w wyniku mutacji co najmniej 5 genów mitochondrialnego DNA.
Geny te biorą udział w syntezie reduktazy tlenkowej NADH lub kompleksu I oraz niektórych tRNA.
Zespół MELAS obejmuje obecność zaburzeń neurologicznych, mięśniowych, nietypowe nagromadzenie kwasu mlekowego w tkankach (wraz ze wszystkimi towarzyszącymi objawami), problemy z oddychaniem, utratę kontroli czynności jelit, nawracające zmęczenie, problemy z nerkami, problemy z sercem, cukrzycę, epilepsję i brak koordynacji.
INNE PATOLOGIE
Według różnych badań naukowych, w chorobach takich jak zespół cyklicznych wymiotów, barwnikowe zwyrodnienie siatkówki, ataksja, choroba Parkinsona i choroba Alzheimera występuje również zaangażowanie mitochondrialnego DNA i niektórych jego mutacji.