Definicja talasemii
Talasemia to genetycznie przenoszona choroba krwi, w której organizm syntetyzuje nieprawidłową formę hemoglobiny.
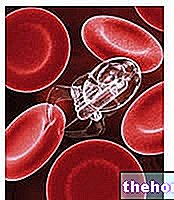
Jak większość ludzi wie, hemoglobina jest białkiem zawartym w czerwonych krwinkach, niezbędnym do transportu tlenu we krwi. U osób cierpiących na talasemię zmutowana forma hemoglobiny powoduje stopniowe, ale nieubłagane niszczenie czerwonych krwinek, aż do anemii.
Ze statystyk medycznych jasno wynika, że talasemia dotyka głównie mieszkańców krajów Bliskiego Wschodu, krajów afrykańskich oraz wszystkich tych, którzy zamieszkują tereny bagienne (nic dziwnego, że talasemia nazywana jest również Anemia śródziemnomorska).
Klasyfikacja i przyczyny
Ze względu na wadliwą podjednostkę białkową (która tworzy hemoglobinę) rozróżnia się dwie formy talasemii, przed przystąpieniem do analizy cofnijmy się, aby wyjaśnić kilka bardzo ważnych pojęć.
Hemoglobina jest nośnikiem par excellence, używanym do transportu tlenu we krwi, składa się z dwóch białek, znanych jako alfa-globulina i beta-globulina.
Talasemia występuje, gdy jeden lub więcej genów kontrolujących produkcję jednego lub obu tych białek jest uszkodzony (zmutowany).
Talasemia jest spowodowana mutacją DNA białek tworzących hemoglobinę: zmiany te mają duży wpływ na fizjologiczną syntezę hemoglobiny i, niszcząc erytrocyty, prowadzą do anemii.
Klasyfikacji talasemii należy dokonać na podstawie dwóch ważnych czynników:
- Liczba zmutowanych genów odziedziczonych po rodzicach
- Rodzaj zaangażowanego białka (hemoglobina alfa lub beta)
Talasemia alfa
W „alfa” postaci talasemii – w której 4 „alfa” podjednostek kulistych hemoglobiny (w chromosomie 16) mogą być zmutowane – zaangażowany jest jeden lub więcej wadliwych genów; każda podjednostka kulista jest wyraźnie zakodowana z genu, dlatego geny, które mogą być zaangażowane, to 4.

Ogólny obraz objawów staje się poważniejszy, gdy zaangażowane są trzy lub cztery geny: w pierwszym przypadku mówimy o „choroba hemoglobiny H„(Z umiarkowanymi lub ciężkimi objawami). Gdy zaangażowane są wszystkie cztery geny, choroba nazywa się alfa-talasemia major: w podobnych sytuacjach noworodek umiera na krótko przed porodem lub wkrótce po nim.
Talasemia beta
Postać beta talasemii, jak można się domyślać, występuje, gdy geny zaangażowane w skład łańcuchów beta są zmutowane (na poziomie chromosomu 11): w tym przypadku mogą dotyczyć tylko dwóch genów. Jeśli tylko jeden gen zostanie zmieniony, określa się to jako beta-talasemia drobna, w którym pacjent skarży się na brak istotnych objawów. Podobnie jak w wariancie alfa, zaangażowanie obu genów tworzących łańcuchy beta hemoglobiny skutkuje jednym beta-talasemia major (lub Anemia Cooleya), która odzwierciedla ciężkie i ciężkie objawy; w tym przypadku jednak objawy zwykle zaczynają się po kilku latach od urodzenia.
Obejrzyj wideo
- Obejrzyj wideo na youtube
Objawy
Więcej informacji: Objawy talasemii
Talasemia jest bardzo poważną chorobą dziedziczną, do tego stopnia, że niektóre jej warianty, takie jak alfa-talasemia major, mogą spowodować śmierć dziecka podczas porodu lub wkrótce po urodzeniu.Niemowlęta z beta-talasemią mogą jednak przeżyć i rozwijać się pierwsze objawy w ciągu kilku lat od urodzenia (ciężka niedokrwistość).
Jeśli tylko jeden gen jest zmieniony, zarówno w postaci alfa, jak i beta talasemii, pacjenci nie skarżą się na żadne znaczące objawy; dopiero dzięki analizie mikroskopowej próbki krwi pobranej od pacjenta dochodzi do nieprawidłowości w kształcie i strukturze erytrocytów, znacznie mniejszej niż norma.
Oprócz anemii u pacjentów z talasemią może wystąpić jeden lub więcej z następujących objawów: zmęczenie, zmiany nastroju (drażliwość), zaburzenia wzrostu, deformacje kości twarzy, żółtaczka, duszność i ciemny mocz.
W przypadkach nasilenia obraz symptomatologiczny pacjenta cierpiącego na talasemię może ulec degeneracji, aż do powstania prawdziwych deformacji kości, zwłaszcza twarzy i czaszki; talasemia może promować „nieprawidłową ekspansję szpiku kostnego, zarówno przez osłabienie masy kostnej, jak i przez ogromne zwiększenie ryzyka złamań kości.
Wśród powikłań talasemii należy również wymienić możliwe nagromadzenie żelaza (hemochromatoza), będące wyrazem zarówno samej choroby, jak i nawracających przetoczeń krwi, których pacjent potrzebuje.
Talasemia często powoduje splenomegalię, czyli nadmierny wzrost objętości śledziony: często ten patologiczny stan kliniczny wymaga splenektomii, chirurgicznego usunięcia narządu.Jak wiemy, śledziona jest ważnym narządem wykorzystywanym do syntezy krwinek oraz przeciwciała, oprócz kontroli infekcji: ich usunięcie wyraźnie sprzyja osłabieniu funkcji obronnej przed atakami bakteryjnymi i wirusowymi, czyniąc podmiot bardziej wrażliwym na infekcje.Należy jednak podkreślić, że sama talasemia również zwiększa ryzyko zarażać się infekcjami: w przypadku wycięcia śledziony w kontekście talasemii szanse na infekcję przesadnie wzrastają.

Diagnoza
Jeśli ojciec i/lub matka są dotknięci talasemią, prawdopodobieństwo przeniesienia choroby na potomstwo jest bardzo wysokie. Przeanalizowaliśmy, że nie wszystkie postacie talasemii zaczynają się od dokładnej symptomatologii od urodzenia: w podobnych sytuacjach, w przypadku podejrzenia talasemii, możliwe jest poddanie chorego szeregu specyficznych testów i badań, mających na celu ocenę diagnostyczną ( takie jak oznaczanie hemoglobiny A2, która jest podwyższona u zdrowych osób z genami beta-talasemicznymi).
Wśród badań fizykalnych, medyczne badanie dotykowe śledziony może czasami wykazać talasemię: jak już wspomniano, splenomegalia stanowi pierwszy sygnał alarmowy dla anemii śródziemnomorskiej. Badania krwi są bardziej szczegółowe i precyzyjne: w próbce krwi z talasemii czerwone krwinki oglądane pod mikroskopem wydają się małe i mają nieprawidłowy kształt. Co więcej, dokładna morfologia krwi pacjenta cierpiącego na talasemię ujawnia ciężką anemię: test ten jest przydatny do oznaczania stężenia żelaza we krwi, wykonywania analizy DNA w celu oceny diagnostycznej choroby i oceny możliwej mutacji „hemoglobiny”. .
Z drugiej strony elektroforeza hemoglobiny ujawnia nieprawidłowy kształt białek przenoszących tlen.
Niektórych wariantów talasemii nie można zdiagnozować za pomocą elektroforezy: w tym przypadku pacjent zostanie poddany testowi „analizy mutacyjnej”, który jest przydatny do wykrywania i potwierdzania talasemii.
Leki i zabiegi
Zobacz też: Leki stosowane w leczeniu talasemii
Ponieważ jest to choroba przenoszona genetycznie, zrozumiałe jest, że - na razie - nie ma leku zdolnego do odwrócenia choroby; jednak możliwe jest kontrolowanie objawów, poprawiając jakość życia pacjenta. Wybór jednego leczenia, a nie innego, zależy od rodzaju talasemii i nasilenia objawów.
W łagodnym wariancie talasemii (w którym np. zmienia się tylko jeden gen) nie są potrzebne żadne leki, ponieważ pacjent nie skarży się na objawy. W takich okolicznościach nadal wskazane jest regularne przeprowadzanie niezbędnych kontroli; Czasami przydatne są okazjonalne transfuzje krwi (szczególnie w przypadku operacji i porodu).
W przypadku umiarkowanych lub ciężkich postaci objawowych podejście do leczenia jest inne i może wymagać częstych transfuzji krwi lub, w ciężkich przypadkach, przeszczepu komórek macierzystych.
- Transfuzje krwi: to podejście terapeutyczne może również powodować poważne komplikacje, ponieważ częste transfuzje mogą sprzyjać patologicznej akumulacji żelaza we krwi (hemochromatoza), co wymaga specyficznego leczenia mającego na celu wyeliminowanie magazynowania żelaza, znanego jako chelator terapeutyczny (z lekami takimi jak Deferasirox i Deferypron). Więcej informacji: przeczytaj artykuł o lekach do leczenia hemochromatozy.
- Przeszczep szpiku kostnego: zarezerwowany dla najpoważniejszych przypadków, w których talasemia powoduje poważne dysfunkcje w organizmie.